
Abstract
Purpose:
Fanconi anemia (FA) is a complex tumor-prone disease defined by an entangled genotype and phenotype. Despite enormous efforts in the last 20 years, a comprehensive and integrated view of the disease is still missing. The aim of this pilot study was to establish whether a global microRNA (miRNA) analysis approach could be helpful in defining aspects in FA phenotype, which might deserve future attention with the perspective to develop miRNA-based therapies.
Methods:
miRNA array were employed to characterize the global miRNA (miRNoma) profile of FA RNA samples with respect to normal samples.
Results:
We report and compare miRNA profile from two FA established cell lines and three FA patients. This analysis reveals that 36 and 64 miRNAs, respectively, are found differentially expressed (>2-fold variation and P < 0.05) in the samples from FA cell lines and FA patients. Overlap of these data results in 24 miRNAs as shared in the two sample populations. Available bioinformatics methods were used to predict target genes for the differentially expressed miRNAs and to perform pathway enrichment analysis.
Conclusions:
Seven pathway results associated with the FA phenotype. It is interesting to note that some of these pathways were previously unrelated to FA phenotype. It might be important to focus on these pathways not previously emerged as dysfunctional in FA to better define the pathophysiological context of this disease. This is the first report of a global miRNA analysis in FA.
Introduction
Fanconi anemia (FA) is a rare inherited bone marrow (BM) failure syndrome that features very high cancer predisposition. Approximately, 20% of the patients have some type of malignancy, 45% of which are leukemias. The high incidence of acute myeloid leukemia and myelodysplastic syndromes is probably due to the very young age of manifestation of the disease. 1
FA is an extremely complex disease. So far, at least 23 complementation groups 2 are associated with the disease. FA displays defects in the DNA repair processes, in the energy and oxidative metabolism, in cytokine expression, cell structure and morphology, and calcium homeostasis. 3,4 Furthermore, FA displays mitochondria functional and structural defects. 5 Despite the enormous increase in the knowledge for FA biological mechanisms during the last 20 years, an integrated view of the metabolic, regulatory, and functional relationships of the disease is still missing, and hence, the characterization of the metabolic defects associated with FA is still increasing. 6,7 In our opinion, the use of global analytical technologies (DNA, RNA, etc.) could help in defining this complex scenario. To address this point, we have chosen to analyze and characterize the microRNA (miRNA) differential expression in a small population of FA samples both from established cell lines and patients.
Mature miRNAs are small noncoding RNA molecules, ∼22 nucleotides in length, ubiquitous in the living world, which act in RNA silencing and post-transcriptional regulation. 8 Over more than 2000 potential human miRNAs have been classified. The relationship between miRNAs and mRNA is degenerated. A single miRNA binds many mRNA sequences and each mRNA sequence is target of many miRNAs. This results in a fine-tuning of gene expression, while it significantly complicates their use as diagnostic and medical tools.
The influence of miRNA expression on the target genes and the connections between the expression of selected miRNAs and the development of various diseases is actually well known as well miRNA signaling involved in cancer development. 9 The development of miRNA microarrays and the global miRNA analysis allow the identification of the miRNAs involved in the carcinogenic process. The definition of the specific set of miRNAs, the miRNA signature, as characteristic in reference to selected tumors, now appears an informative diagnostic tool. 10 Actually, tumors originated from various tissues can be classified solely on the basis of their miRNA profiles. 11
Concerning FA, research “omics” techniques have been somehow neglected. Few reports deal with gene microarrays, 12 –14 proteomics, 15 metabolomics, 16 and lipidomics. 17 Concerning miRNAs, while very few reports have dealt with this issue, significant information has been reported in a study by Rio et al. 18 and Suresh et al. 19 The study of Rio et al. 18 reported analysis on the expression of 157 miRNAs in lymphoblastic cell lines and peripheral blood cells from FA patients and showed that three miRNAs (hsa-miR-133a, hsa-miR-135b, and hsa-miR-181c) were downregulated in FA hematopoietic cells. Among these miRNAs, the authors concentrate on hsa-miRNA-181c, which targets tumor necrosis factor (TNF) and interleukin-1, two cytokines elevated in FA patients, a consolidated feature of FA disease. The authors hypothesized that this finding may likely open up new strategies in the treatment of hematologic dysfunction in FA patients based on miRNA regulation. The report of Suresh et al., 19 on the other hand, identified the miR-302 cluster as a potential regulator of FANCD2. miR-302 is involved in the maintenance of cell totipotency and self-renewal in human and mouse embryonic stem cells. Overexpression of miR-302 plays a critical role in the regulation of FANCD2 monoubiquitination, resulting in characteristic defects in DNA repair within cells.
In conclusion, the regulatory role of miRNA in FA appears promising for the identification of new mechanisms of regulation or the definition of mechanisms already known. We consider important to carry on the study of the role of miRNAs in FA. Eventual limitation to this approach could limit an appropriate knowledge of FA molecular regulation and wiring.
Materials and Methods
Patients, controls, and cell lines
The use of FA patient samples was under approval (Ethics Committee of G. Gaslini Children's Institute, Genoa, Italy). Control samples were taken from healthy voluntary donors (HD samples). Informed consent was obtained from patients, controls, and/or their relatives according to the procedures (Ethics Committee of G. Gaslini Children's Institute).
Primary bone marrow mononuclear cells (MNCs) were isolated using Ficoll-Paque Plus (GE Healthcare Biosciences, Piscataway, NJ) from three FA patients and three BM donors (HD). Two Fanc-A Lymphoblast cell lines were obtained from the “Cell Line and DNA Biobank from Patients affected by Genetic Diseases” (G. Gaslini Institute)—Telethon Genetic Biobank Network (Project No. GTB07001). 20 Both of these Fanc-A Lymphoblast cell lines were corrected with S11IN retrovirus carrying on FANC-A wt gene 21 to generate the FA-corrected cell lines (mutant FA genes silenced). Cells were grown at 37°C in RPMI supplemented with 10% fetal calf serum with glutamine and antibiotics.
RNA extraction and analysis
Total RNA from MNCs and lymphoblasts was extracted. 22 RNA quantity and quality were analyzed (Nanodrop, ND-1000; Scientific Thermofisher) by calculating 260/230 and 260/280 absorbance ratios. The Qubit quantitation assay was used as performed in Qubit™ 3.0 Fluorometer (Life Technologies, Gent, Belgium).
Sample labeling and miRNA array hybridization
For evaluating the expression of miRNAs, we use the seventh generation miRCURY LNA™ microRNA Array (Exiqon), which contains 3100 capture probes covering human, mouse, and rat miRNAs. In particular, this microarray analyzes the expression of 1928 human miRNAs. One microgram RNA from each sample was labeled with Label IT® miRNA Labeling Kits, Version 2 (Mirus Bio, WI), following the standard protocol. Total RNA was mixed with 10 μL 10 × labeling buffer, 4 μL Label IT reagent (containing Cy 3 or Cy 5 fluorescent tracers), and water to 86 μL. The samples were incubated at 36°C for 1 hr and the reaction was stopped by adding 10 μL Stop Reagent. Labeled samples were purified on a chromatographic column and eluted in 25 μL elution buffer. Then, Hybridization Solution (EXIQON, Vedbaek, Denmark) was added and the resulting mixture denatured at 65°C for 3 min. The labeled mix was transferred to the microarray and covered with coverslips. The hybridization was performed in GlassArray Hybridization Cassettes (Invitrogen Ltd., Paisley, United Kingdom) in a water bath at 37°C for 16 hr and a wash sequence was performed. The array was dried by centrifugation and laser scanned (ScanArray; PerkinElmer, Waltham, MA) to record fluorescent signals produced by each spotted probe effectively hybridized with the corresponding miRNA.
Data analysis
The microarray data were processed by GeneSpring software. The local background was subtracted, data log-transformed, and normalized per gene/chip mean. Data overall variability, as related to FA status, was examined by box plot analysis, scatter plot analysis, hierarchical cluster analysis, and principal component analysis. Individual miRNAs modulated by FA were identified by volcano plot analyses.
Validation by real-time qPCR
Microarray data for selected miRNAs have been validated by qPCR. qPCR reactions and identification of primer sequences have been performed by using the miRCURY LNA™ Universal RT microRNA PCR Exiqon (Woburn, MA). First-strand cDNA was synthesized from selected miRNAs by using 5 × reaction buffer (2 μL), nuclease-free water (5 μL), a reverse transcriptase mix (1 μL), and template total RNA (5 ng/μL) in a total volume of 10 μL. The reaction mixture has been incubated for 60 min at 42°C and 5 min at 95°C, and cooled at 4°C to stop the reaction. Real-time PCR amplification was performed by Rotor-Gene (Corbett Research, Mortlake, Australia). For each tested miRNA, a first-strand cDNA template (4 μL) was added with Exilent SYBR Green master mix (5 μL) and PCR primer/enzyme set mix (1 μL) to a total volume of 10 μL. DNA polymerase was activated by heating at 95°C for 10 min (hot start reaction); 45 amplification cycles were performed at 95°C for 10 sec, and thereafter at 60°C for 1 min. The specificity of the obtained amplicons was tested by analyzing melting curves.
Bioinformatics analysis
In the last few years, several bioinformatics tools have been developed to predict miRNA gene targets. These softwares rely on a combination of specific base-pairing rules and conservational analysis to score possible miRNA base-pairing sequences in the recognition sites of putative target genes. Several analytical procedures and experimental information have contributed to improve the miRNA gene target prediction algorithms dedicated to this purpose.
11,23,24
Each miRNA differentially expressed between the FA and the controls was individually studied through the available literature from in silico or experimental studies and combining the data accessible from web resources [miRMap 2.0 (
Essentially, the MiRBase web resource, which is a biological database that acts as an archive of miRNA sequences and annotations, was used to get basic information on the predicted gene targets for single miRNA and the target gene functions. DIANA-miRPath 25 was used to gather information associated with pathway-related miRNA combinations. miRNA combinations associated to diseases and cancers were obtained through the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, which represents diseases as perturbed states of the biological system.
Results
Data analysis
The analysis of miRNA profile by scatter plot (Fig. 1A) revealed that multiple miRNAs are twofold upregulated or downregulated in FA compared to FA-corrected samples. These miRNAs are identified as dots located outside the twofold diagonal variation interval (green lines). Volcano plot analysis identified 36 miRNAs as differentially expressed (>2-fold variation and P < 0.05) in the samples from FA cell lines in comparison to FA-corrected samples (Fig. 1B). Analysis of samples from FA patients resulted in the identification of 64 miRNAs as differentially expressed (>2-fold variation and P < 0.05) in comparison to the HD samples (Fig. 1C).
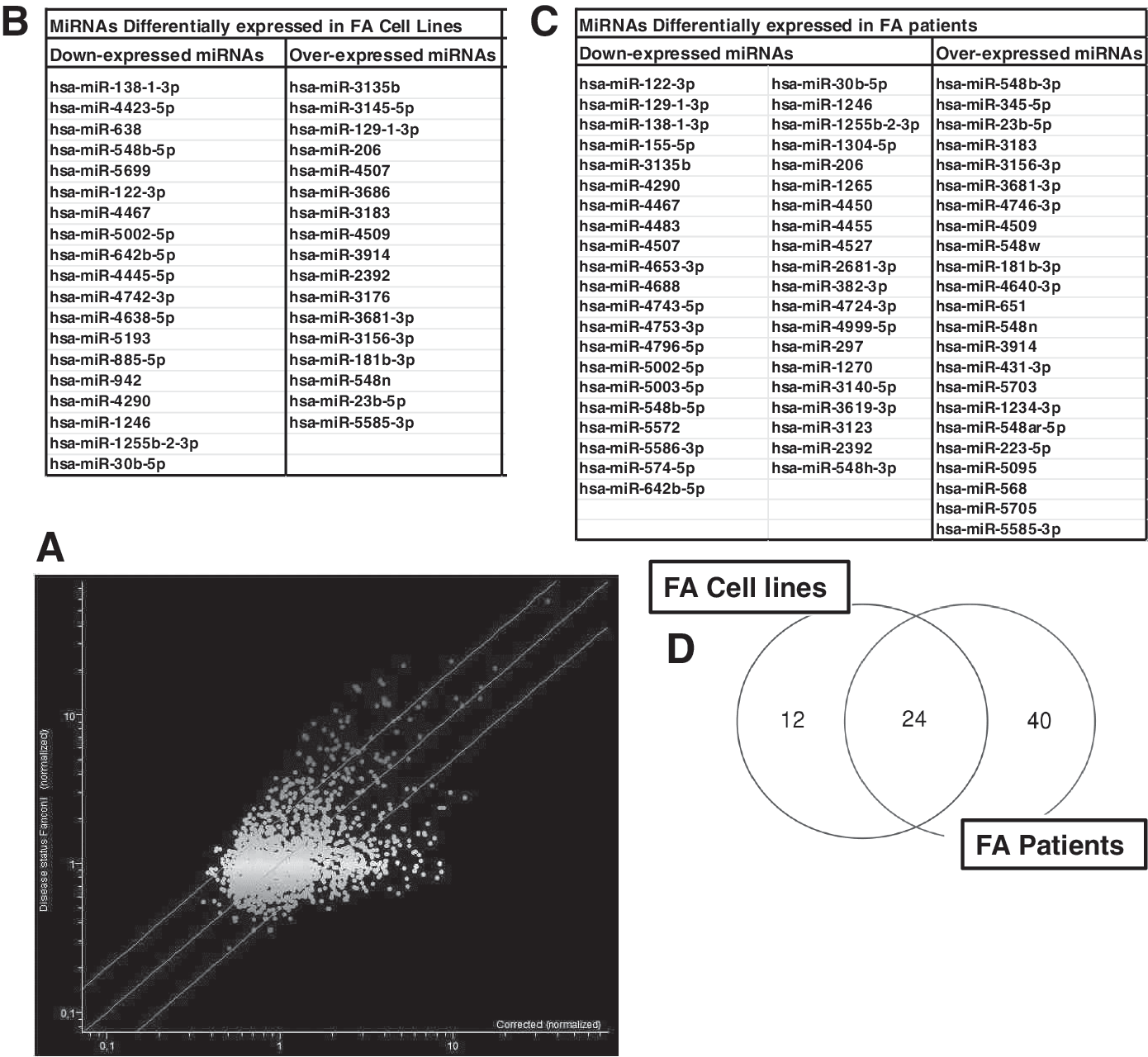
Data from the analysis of miRNA profile in FA cells and patients compared to FA-corrected cells and donor controls.
Superimposing the results obtained in the analyses between cell lines and patients, 24 miRNAs are found to be shared in the two populations (Fig. 1D; Table 1). The modification of miR-30b, miR-138, and miR-181 was validated by qPCR. The fold variations observed were 0.55 ± 0.18 for miR-30b (downregulation), 0.42 ± 0.10 for miR-138 (downregulation), and 2.24 ± 0.46 for miR-181 (upregulation), consistent with the results obtained by microarray.
Differentially Expressed MicroRNAs in Fanconi Anemia Cell Lines and Patients
miRNA, microRNA.
Data mining
The 24 miRNAs in whole were used to perform the global analysis. DIANA and KEGG web resources were used for this purpose. 26 Applying a significance score of <0.05, 16 among the original 24 miRNA reports link to seven pathways (Fig. 2).
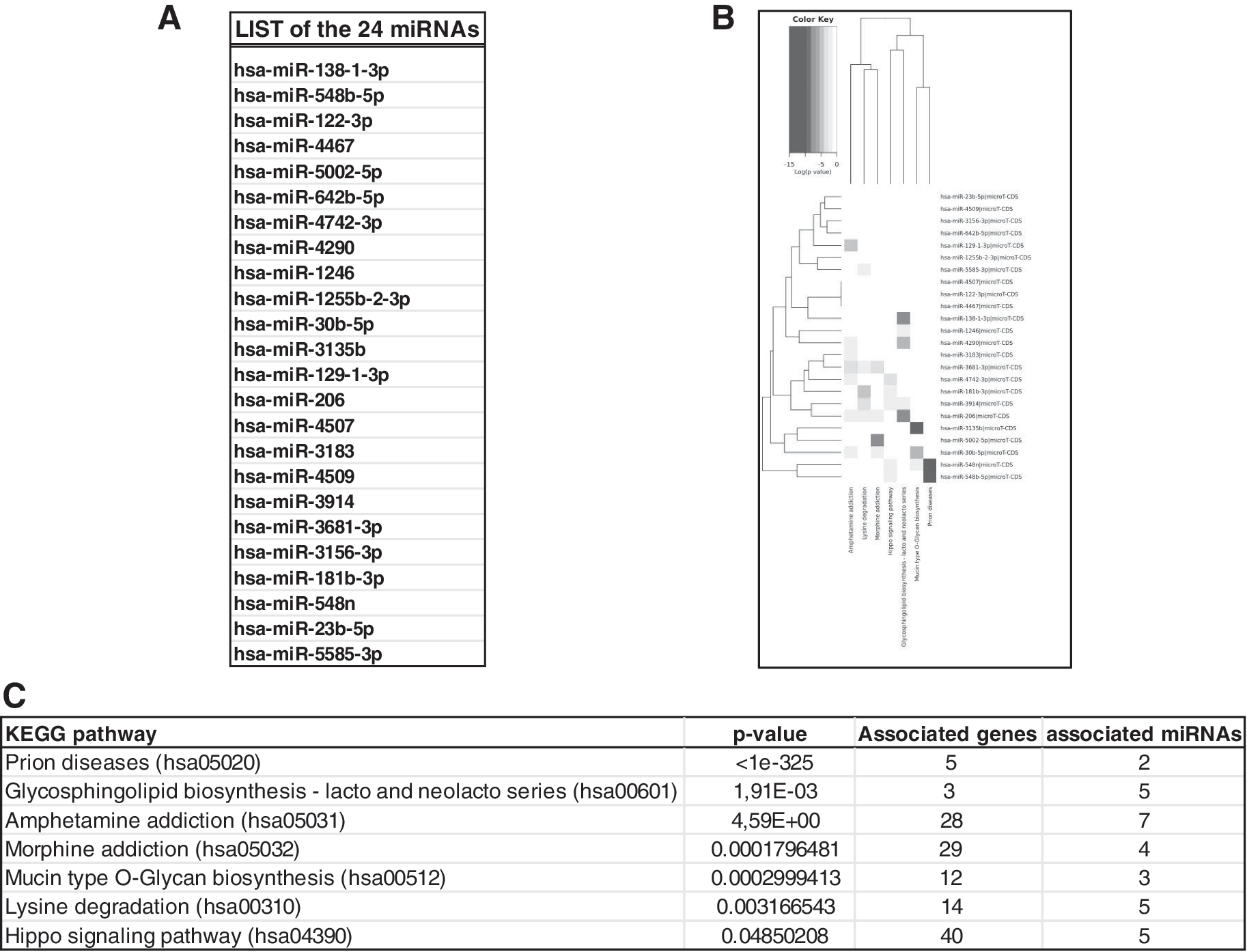
Global analysis of the 24 miRNAs differentially expressed in FA samples.
Discussion
This study aims to identify the carcinogenic potential-related molecular mechanisms in FA cells by analysis of the global miRNoma. This is the first study relating to the global miRNoma in FA. The KEGG pathway enrichment analysis indicated seven modules primarily associated with prion disease, glycosphingolipid (GSL) biosynthesis—lacto and neolacto series, amphetamine addiction, morphine addiction, mucin-type O-glycan biosynthesis, lysine degradation, and Hippo signaling pathway. It is interesting to note that at least some of the pathways emerging in this analysis were previously unrelated to FA phenotype, while their involvement in this analysis might get directions and hints for future research.
Indeed, the association between the prion pathway and FA may exquisitely relate to structural problems emerging in the replications of DNA structures that present expansions as G4 motifs typical in telomeric repeats. 27 Mechanistically, a G4 structure may cause the stalling of the replication machinery and promote the formation of chromosomal breaks. G4 structure replication needs assistance by specific helicases. The association between G4 and FA emerged in a study on dog-1, an ortholog of the human gene FANCJ 28 involved for the maintenance of epigenetic stability. The promoters of the XPD and XPB regions are enriched with G4 structures, and mutations in these regions are associated with three genetic diseases (Xeroderma pigmentosum, trichothiodystrophy, and Cockayne syndrome), which share several homologies with FA. 29,30
The pathway associated with GSLs, biotin, and mucin-type O-glycan in FA might relate to a dysmetabolism of fatty acids and glucide-associated membrane components. A consistent literature on the mitochondrial related and energy dysfunction in FA 3 –5 is now available. Recently, 6 defects in the lipid metabolism are also being addressed.
Biotin deficiency is not only linked to mild clinical symptoms such as hair thinning or skin rash but is also associated with increased cancer resistance through nuclear factor kappa-light-chain-enhancer of activated B cells, and it regulates TNF-alpha production in murine macrophages. 31
Mucin-type O-glycosylation is an evolutionarily conserved protein modification of membrane-bound and secreted proteins. Aberrations in O-glycosylation are associated with diseases related to protein secretion, stability, processing, and function. 32
The T, Tn, and Lewis antigens are tumor-associated carbohydrate antigens. In breast cancer, these antigens are associated with a poor prognosis and a reduced survival, and their presence may alter the cellular susceptibility toward an invasive and metastatic potential. 33
Finally, alterations in the pathways associated with lipid and glucide metabolism have recently been related to hematoppoietic stem and progenitor cells (HSPC) self-renewal and differentiation potential. 34,35
Still, concerning fatty acid metabolism, GSL metabolism deserves further attention. Indeed, GSL dysmetabolism is related to squamous cell carcinomas whom FA patients display high rate of predisposition with relevance to the keratinocyte-based carcinomas of the head and neck [head and neck squamous cell carcinoma (HNSCCs)]. A lipidomic study 17 has recently acknowledged this defect in FA. While the authors discuss this defect in relationship to FA's DNA repair defect, the altered lipid metabolism can be seen as resulting from the metabolic impairment of OXPHOS function in FA cells, which determines a slowing of the Krebs cycle flux, causing the AcCoA accumulation, which finally determines a lipid accumulation. 4 –6
HNSCC is a leading type of cancer worldwide with limited therapeutic options and remarkable negative effects. A better knowledge of the molecular mechanisms at play in HNSCC and the involvement of the FA pathway in this pathological context might be helpful in their clinical management.
Finally, the link to the Hippo signaling pathway is again potentially very interesting in the context of the FA phenotype. Indeed, this pathway is involved in restraining cell proliferation and promoting apoptosis during organism development. The pathway has also critical role in stem cell self-renewal and expansion. The Hippo pathway has become increasingly significant in the study of the unchecked proliferation processes occurring in cancer development.
The core components in the Hippo pathway are the yes-associated protein (YAP) and transcription coactivator with PDZ binding motif (TAZ), proteins activated downstream events damaging DNA. The pathway appears involved in the protection of DNA from damage and in genome stability, 36 and links extracellular signaling with nuclear transduction. 37 Interestingly, the Hippo pathway maybe seen as a rescue pathway where FA is defective. Indeed, a major role has recently emerged in the context of breast cancer metastasis. 38
The role of the other pathways that come out in the enrichment analysis (amphetamine addiction, morphine addiction, and lysine degradation) seems less obvious in the context of the FA disease. Likely, these potential biases might be overcome in a larger study. Further information concerning each of the miRNA differentially expressed in this study is summarized in the Supplementary Table S1.
This study finally evidences that an altered expression of several miRNAs in FA might be associated with traits of the disease only, in part, already acknowledged. Our analysis highlights a possible role in FA of pathways associated with defects in membrane and fatty acid metabolism, an issue that has not yet been satisfactorily covered in FA research and only very recently being acknowledged. 6,39,40
Pathway analysis may be regarded as a first choice to gain insight in the biology of differentially expressed genes and proteins and their regulation. This approach consistently permits to reduce the complexity of the systems under study and has great explanatory and forecast power in data manipulation. 26 Still, several limitations have to be overcome in this approach. It is necessary to implement the informatics tools dedicated to these analysis, and recognize that our knowledge on the RNA is still limited and in constant evolution. 41
In conclusion, this pilot study is the first report on a global analysis of miRNAs in FA. Despite the limited quantity of samples examined, we believe that the information obtained are significant and adequate to the phenotypic context that emerges in the biochemical and molecular studies on FA. This report adds and completes a picture of specific pathophysiological issues of the disease. The study recommends the improvement of genome-oriented studies in the research on Fanconi's anemia.
Ethical Statement
The study has been performed in accordance with the ethical standards of the Declaration of Helsinki and its later amendments.
Informed Consent Statement
Informed consent was obtained from all individual participants included in the study.
Footnotes
Authors' Contributions
P.D.: experimental design, experimental work, sample preparation, sample contribution, data analysis, discussion, and article authorship; E.C.: experimental design, sample contribution, discussion, and article authorship; M.G.L.: experimental work and sample preparation; A.P.: article authorship; P.C.: experimental design, experimental work, sample preparation, and article authorship; C.D.: sample contribution, discussion, and article authorship; S.R.: experimental design, data analysis, discussion, and article authorship; D.C.: experimental design, data analysis, discussion, and article authorship; A.I.: experimental design, experimental work, data analysis, discussion, and article authorship.
Acknowledgments
P.D. acknowledges Associazione Italiana per la Ricerca sull'Anemia di Fanconi (AIRFA), Fondi 5x1000 2013 to IRCCS AOU San Martino—IST (to date renamed as Ospedale Policlinico San Martino, Genova).
Author Disclosure Statement
No conflicting financial interests exist.
Supplementary Material
Supplementary Table1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.