
Abstract
Heparin-binding EGF-like growth factor (HB-EGF) is an EGF family member that interacts with epidermal growth factor receptor (EGFR) and ERBB4. Since HB-EGF was first identified as a novel growth factor secreted from a human macrophage cell line, numerous pathological and physiological functions related to cell proliferation, migration, and inflammation have been reported. Notably, the expression of HB-EGF is sensitively upregulated by oxidative stress in the endothelial cells and functions for auto- and paracrine-EGFR signaling. Overnutrition and obesity cause elevation of HB-EGF expression and EGFR signaling in the hepatic and vascular systems. Modulations of HB-EGF signaling showed a series of protections against phenotypes related to metabolic syndrome and advanced metabolic diseases, suggesting HB-EGF as a potential target against metabolic diseases.
Introduction
Heparin-binding EGF-like growth factor (HB-EGF) is a primary protein in the macrophage cell supernatants that interacts with the heparin column. 1 Later, it was shown to be a member of the EGF family that interacts with epidermal growth factor receptor (EGFR) and ERBB4. 2 –4 The HB-EGF-EGFR pathway induces multiple signaling pathways depending on cell types, including ERK1/2, PI3K-Akt, PLC-γ, and STATs. 5 HB-EGF is expressed as a type I transmembrane protein on the cell surface. 4,6 A list of metalloproteinases (MPs) activates the HB-EGF for auto- or paracrine-signaling. 7 Mature HB-EGF still contains a heparin-binding motif; thus, it interacts with cell surface proteoglycan. 1,6,8,9 Membrane-tethered HB-EGF can form a complex with neighbor cell EGFR for juxtacrine signaling. 4,10 Oxidative stress inducers sensitively upregulate the HB-EGF transcription in the endothelial cells. 11,12 HB-EGF expression in bone marrow stromal cells increases the proliferation of the hematopoietic stem cells and progenitor cells (HSPCs). 13,14 Recent research results indicate that increased myeloid cell production in bone marrow is a significant feature in the development of metabolic diseases. 15,16 The regulatory function of HB-EGF in hematopoiesis may be a connection of oxidative stress with induction of low-grade inflammation under metabolic stress environments. We also summarized recent reports on the potential of HB-EGF targeting against the advancement of metabolic diseases (Table 1). Different approaches of HB-EGF targeting induced a list of protective phenotypes in the animal and human studies, suggesting HB-EGF as a potential target for therapeutic purposes.
Functions of Heparin-Binding EGF-Like Growth Factor Related to Phenotypes of Metabolic Syndrome and Diseases
EGFR, epidermal growth factor receptor; HSPCs, hematopoietic stem and progenitor cells; LSECs, liver sinusoidal endothelial cells; VLDL, very-low-density lipoprotein.
Effects of the HB-EGF gene deletion and overexpression in metabolic disease phenotypes
Germline deletion of HB-EGF in systemic or vascular endothelium caused cardiac hypertrophy with gross enlargement of ventricular chambers of the heart 17,18 ; however, the postnatal induction of HB-EGF gene deletion did not induce the deleterious effects. 19 –21 Although the basal level of HB-EGF expression in the hepatocytes is low, hepatocyte-specific HB-EGF gene deletion induced an increase of inflammation and fibrosis in the liver. 22 –24 Interestingly, HB-EGF overexpression also enhanced the induction of liver damage, 25 suggesting that a physiological range of HB-EGF expression is essential for a healthy liver. The endothelial cell-specific HB-EGF gene deletion protected against both diabetic- and angiotensin-II (AngII)-induced renal disease phenotypes in animal models. 19,26 The podocyte-specific HB-EGF gene deletion also induced protection against acute renal disease in a mouse model. 21
HB-EGF mediates EGFR transactivation under oxidative stress
Reactive oxygen species (ROS) formations and activation of MPs are involved in EGFR transactivation in the cells and tissues under oxidative stress environments. 3,12,27,28 Numerous reports indicated a crucial role of HB-EGF in the EGFR transactivation by oxidative stress inducers, 3,29,30 including AngII, 31 catecholamines, 32 and lipid oxidation products. 33,34 Differently to the canonical ligand-activated signaling, the stress-induced EGFR transactivation showed a low-level but prolonged signaling with minimal internalization or desensitization of EGFR. 30 Although there are >10 ligands for EGFR, the HB-EGF almost exclusively mediates EGFR transactivation under various stress environments, 4,35 suggesting that the HB-EGF is a specialized and conserved mediator for the connection of oxidative stress with cell signaling. 3,36 A report indicated that the EGFR was activated in <1 min by endothelin-1, suggesting a mechanism of transcription-independent fashion for the transactivation process. 37 In addition, the upregulation of HB-EGF expression would contribute to the sustained EGFR signaling. 12 The transcription of HB-EGF is mainly controlled by a stress signal associated activator protein-1 (AP-1) transcriptional factor in the cells. 11,38 Oxidative stress inducers also upregulated a list of MPs involved in the HB-EGF processing on the cell surface of endothelial cells. 7,12,34
The obese individuals showed accumulations of oxidation products of phospholipid in the adipose and skeletal muscle tissues. 39 –41 The oxidation products activated AP-1 in the endothelial cells, as demonstrated by a recent Chip-Seq analysis. 42 AngII is a well-known oxidative stress inducer via ROS production through the NADPH oxidase (NOX) system in the vascular smooth muscle cells. 43,44 Correspondingly, AngII induces EGFR transactivation in the cells via HB-EGF mediation. 7,45,46 The EGFR transactivation was an underlying mechanism for the proliferation and migration of vascular smooth muscle or glomerular mesangial cells under the stress conditions. 35,45,47,48 Unsaturated lysophosphatidic acid also induced intimal thickness in the carotid artery via HB-EGF-mediated EGFR transactivation. 29,30,49,50
Role of HB-EGF in the Development of Metabolic Syndrome and Low-Grade Inflammation
The metabolic syndrome is a cluster of metabolic dysfunctions, including central obesity, insulin resistance, dyslipidemia with a manifestation of hypertriglyceridemia, and hypertension. 51 –53 Downregulation of high-density lipoprotein (HDL) and production of small dense low-density lipoprotein (sdLDL) are frequently associated because of the enzyme activity of cholesteryl ester transfer protein, specifically in humans. 53,54
Hyperlipidemia, particularly hypercholesterolemia, is closely associated with the proliferation of HSPCs and the production of myeloid cell progenitors in the bone marrow. 15,55 –57 Under obesity, there are increases in bone marrow-derived monocytes in the bloodstream (monocytosis) and accumulation of proinflammatory macrophages in the adipose tissue. 58 There is also an increase in the local proliferation of macrophages in the atherosclerotic lesion. 59
Role of HB-EGF in the development of low-grade inflammation
The oxidation products of phospholipids induced the upregulation of HB-EGF in the vascular wall, causing inflammatory responses. 60,61 Also, the products of phospholipid peroxidation upregulated the expression of inflammatory cytokines (IL-8 and MCP1/CCL2) and cell adhesion molecules (e.g., ICAM1) in the endothelial cells, leading to the recruitment of bone marrow-derived monocytes into the subendothelial space. 62,63
Under homeostatic conditions, the liver sinusoidal endothelial cells (LSECs) effectively clear any harmful oxidants in circulation, such as oxidized LDL particles (Ox-LDLs) and advanced glycation end products (AGEs). 64 –67 However, a sustained influx of oxidants overproduced in the systemic or splanchnic circulation may induce saturation and activation of the endothelial cells. 68 The saturation of LSECs leads to the activation of extrahepatic endothelial cells and chronic inflammation by prolonged exposure to proinflammatory oxidants.
Krampera et al. demonstrated that HB-EGF is a crucial regulator for the self-renewal of hematopoietic stem cells (HSCs) in the bone marrow. 13,14 The coordination of HB-EGF and CXCL12/SDF-1 in the hematopoietic niches is a determinant for stem cell proliferation and blood cell production in the bone marrow. 14 Various exogenous and endogenous stress inducers, including phorbol myristate acetate and tumor necrosis factor-alpha, upregulated HB-EGF gene expression in the bone marrow. Bone marrow sinusoidal endothelial cells also effectively endocytose oxidants, including minimally oxidized LDLs and AGEs. 64,67 The upregulation of HB-EGF expression in the sinusoidal endothelial cells may lead to the proliferation of HSPCs and the production of myeloid progenitor cells. 69 The endothelial permeability also increased by oxidants, 70 which would lead to the increased mobilization of the HSPCs and immune cells from the bone marrow tissues into circulation. 71
Role of HB-EGF in the development of insulin resistance
Insulin resistance is a central phenotype of metabolic syndrome and is frequently associated with systemic oxidative stress and low-grade inflammation. 53,72 HB-EGF was shown to be involved in the development of insulin resistance by oxidative stress inducers, including endothelin-1, thrombin, and 5-hydroxytryptamine (serotonin) in the adipocytes and skeletal muscle cells. 73 Obesity is a risk factor for the accumulation of ROS in adipose, skeletal muscle tissues, and circulation. 39,40,51 A list of phospholipid peroxidation products also induced insulin resistance in the primary culture of adipocytes and skeletal muscle cells. 41,74 –76 The obese adipose tissue is enriched with proinflammatory M1 macrophages. 39 Concordantly, the HB-EGF gene expression is upregulated in adipose tissue in obese persons. 77 Adiponectin is an established adipokine that is a potent insulin sensitizer. 78,79 The adiponectin level inversely correlated with systemic oxidative stress and visceral obesity. 51,80 Intriguingly, adiponectin directly interacts with HB-EGF for sequestering, 81 –83 which may partly explain the anti-atherogenic and anti-inflammatory functions of adiponectin. 84
The role of HB-EGF in the induction of dyslipidemia
Dyslipidemia, as manifested by hypertriglyceridemia and the reduction of HDL, is the earliest event of metabolic syndrome in obese people. 85 Hepatic very-low-density lipoprotein (VLDL) overproduction is a common feature of hypertriglyceridemia in obese individuals. 53 HB-EGF is mainly expressed in the LSECs in the liver tissue. 86,87 The antisense oligonucleotide (ASO) with a phosphorothioate modification is effectively uptaken by LSECs with effective induction of target gene silencing in the cells. 88 –91 A recent report showed that the HB-EGF ASO administration induced a competent downregulation of circulatory triglyceride (TG) levels by suppressing hepatic VLDL production in a mouse model. 92 Under the obesity condition, the elevated HB-EGF expression in the LSECs may enhance VLDL production in the hepatocytes via a paracrine mechanism. As shown in several cancer cell types, the EGFR pathway may activate the sterol regulatory element-binding protein 1c (SREBP-1c) pathway for the increase of lipogenesis in the hepatocytes. 93,94
The Role of HB-EGF in the Progress of Metabolic Disease Phenotypes
Role of HB-EGF in the development of atherosclerosis
The elevation of TG-rich VLDL particles in circulation is an independent risk factor for the development of atherosclerosis and coronary artery disease. 95,96 Particularly, VLDL remnant and LDL particles derived from VLDLs have optimal sizes for the infiltration into the vascular wall. 62 In addition, the modification of the lipoprotein particles induced self-aggregation in the subendothelial space, 97 which are associated with enhanced scavenging by the macrophages in the subendothelial space. 62 The bioactive components of minimally modified LDL particles, oxidized phospholipids (Ox-PLs), increased gene transcription of HB-EGF and MPs like a disintegrin and metalloproteinase 17 (ADAM17) in the vascular endothelial cells 34,63 (Fig. 1). There were positive associations of HB-EGF content in the vessel wall with the intensity of atherosclerosis in hyperlipidemic mouse and human vessels. 61,98,99 AngII infusion, which elevates HB-EGF-EFR signaling, significantly increased atherosclerosis and aneurysm in hyperlipidemic animal models via HB-EGF upregulation in the vessel wall. 7,100,101 Small-molecule inhibitors of EGFR also showed protection against atherosclerosis and aneurysm in hyperlipidemic animal models. 102,103
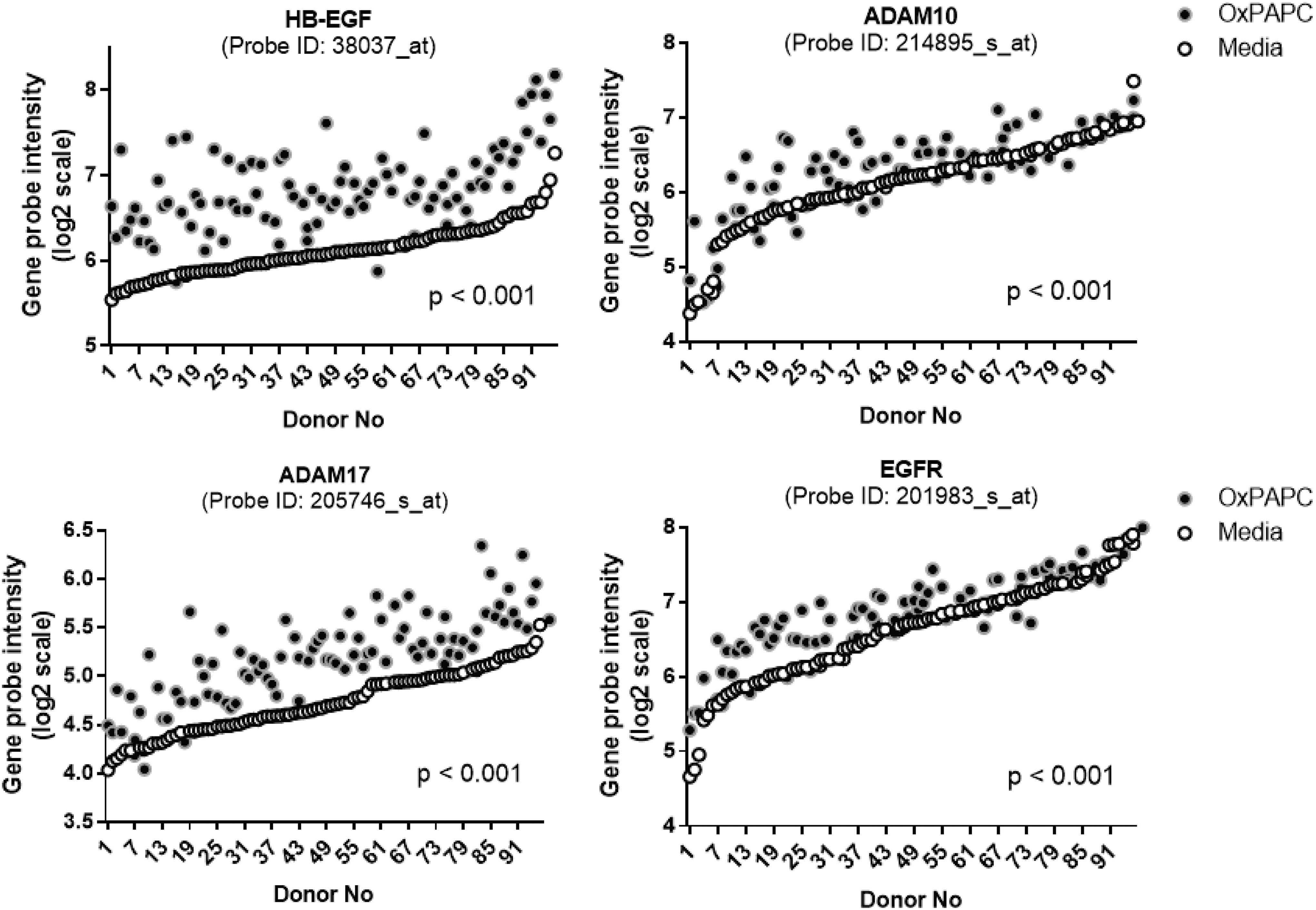
Lipid peroxidation products increased the expression of genes involved in the HB-EGF-activated EGFR pathway in the vascular endothelial cells. The human aortic endothelial cells isolated from 96 different human donors were treated with vehicle and oxidized phospholipid (Ox-PAPC, 50 μg/mL) for 4 hr. The HB-EGF, ADAM10, -17, and EGFR transcript values from the microarray dataset of the donor cells were plotted by order of basal transcript levels. The openly available dataset was reanalyzed (NCBI GEO:
Role of HB-EGF in the development of hepatic inflammation
HB-EGF is mainly expressed in the sinusoidal endothelial cells in the liver. 86,87 Different from the endothelial cells in the other tissues of the body, the LSECs are the scavenger endothelial cells as gatekeepers of the body. 68,104 LSECs effectively scavenge oxidant wastes, including modified (heavily- or minimally oxidized) LDL particles and AGEs, which are significantly enriched in the splanchnic circulation of centrally obese individuals. 51,66,105,106 The sustained influx of oxidants into the hepatic sinusoidal lumen causes the saturation and activation of LSECs for the induction of inflammatory cytokine expression. 68,107,108 The upregulation of HB-EGF in the activated LSECs would induce paracrine EGFR signaling in the hepatocytes for the stimulation of VLDL production. The HB-EGF expressed on and released from the LSECs may cause dyslipidemia and low-grade inflammation in the liver (Fig. 2 for the schematic diagram for events related to chronic inflammation and dyslipidemia induced by the overnutrition). The saturation of the LSECs would also induce extrahepatic endothelial cell activation, causing low-grade systemic inflammation.
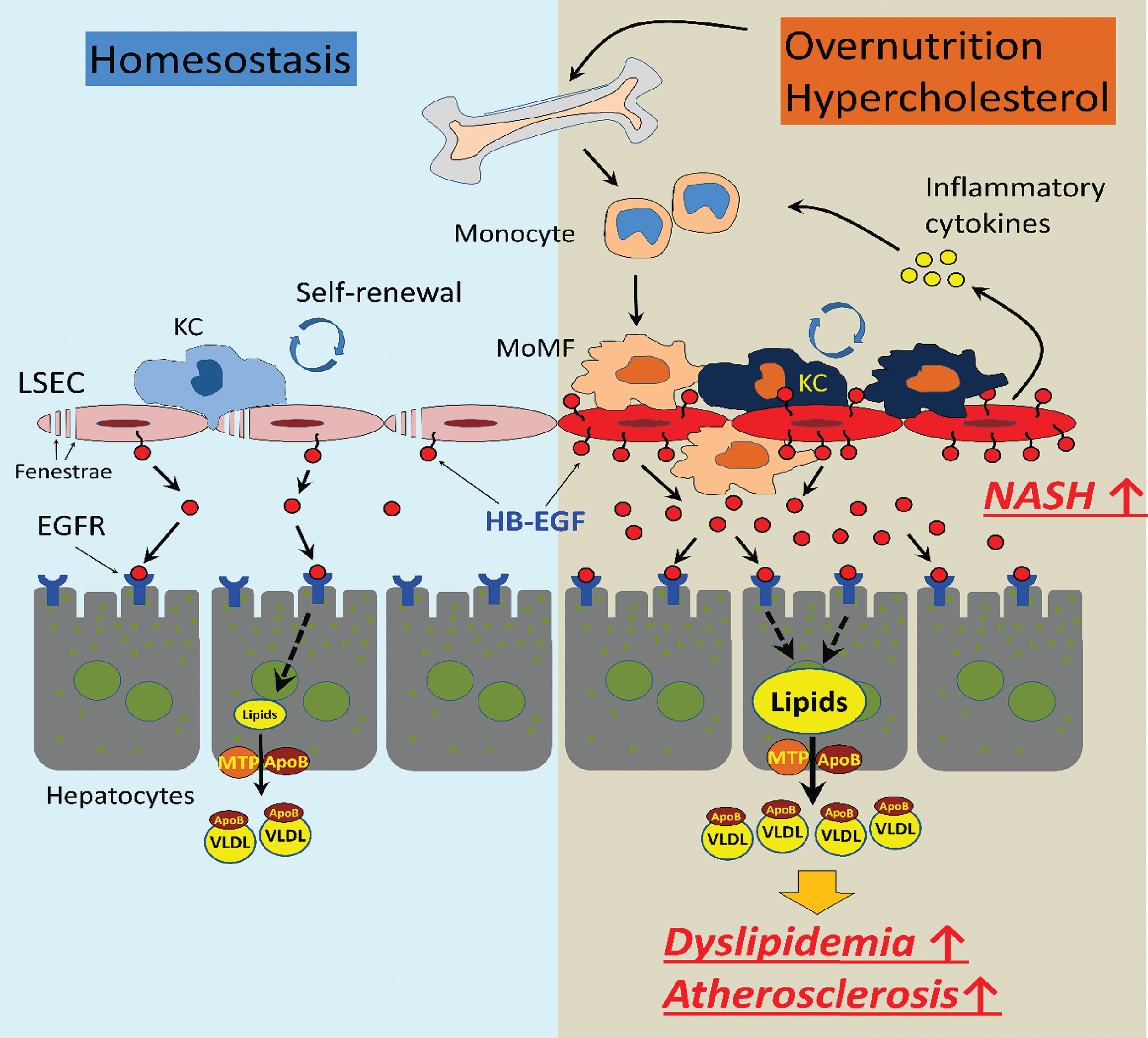
The role of HB-EGF in the regulation of the hepatic inflammation under overnutrition and obesity. In homeostatic condition, HB-EGF is constitutively expressed in and released from the LSECs for the paracrine EGFR signaling in the hepatocytes for basal VLDL production. The oxidative stress associated with nutrition excess and visceral obesity increased the production of harmful oxidants, including oxidized LDL particles in circulation. The oxidants induce saturation and activation of LSECs and upregulation of HB-EGF expression, which may cause extrahepatic endothelial cell activation and recruitment of bone marrow-derived monocyte recruitment and hepatic VLDL overproduction. The elevation of the circulatory cholesterol induces increased proliferation of the myeloid progenitor in the bone marrow, leading to monocytosis and low-grade inflammation. KC, Kupffer cell; LDL, low-density lipoprotein; LSECs, liver sinusoidal endothelial cells; MoMF, monocyte-derived macrophage; MTP, microsomal triglyceride transfer protein; VLDL, very-low-density lipoprotein. Color images are available online.
Pathological role of HB-EGF in renal diseases
Previous reports indicated that HB-EGF-mediated EGFR signaling is involved in the pathology of renal diseases. 21,43,48,109 –112 HB-EGF mediates the disease phenotypes associated with AngII signaling in the kidney. 43 The HB-EGF-mediated EGFR transactivation signaling mediates the fibronectin and transforming growth factor-beta upregulation in renal mesangial cells induced by AngII or high glucose. 47,48 Bollee et al. demonstrated that the podocyte-specific HB-EGF gene deletion or the administration of EGFR blocker induced protections against rapidly progressive glomerulonephritis in a mouse model. 21 The endothelial-specific HB-EGF deletion also induced a series of protection against renal disease induced by AngII, unilateral nephrectomy, 19,113 –115 or ischemic reperfusion. 116 In contrast, the administration of recombinant HB-EGF downregulated glomerulus filtration in the animal model of glomerulonephritis. 20 Because HB-EGF is abundantly expressed in the epithelial cells in distal tubules, there were significant increases in soluble HB-EGF content in the urines under renal disease conditions. 116 Overstreet et al. showed that renal tubular HB-EGF overexpression caused the development of renal fibrosis. 112
The HB-EGF as a Potential Target Against Metabolic Diseases
The accumulation of lipid peroxidation products (e.g., Ox-PLs) in circulation appears to be a risk factor for the development of metabolic syndrome associated with central obesity. 51,52,117,118 The inhibition of ROS production or the neutralization of the oxidants is a possible approach for the protection against metabolic syndrome and disease phenotypes. 119 Considering the upregulation of the HB-EGF in the endothelium by oxidative stress, the HB-EGF targeting or modulation of the HB-EGF signaling could be another option for treatment. The clinical application of HB-EGF modulators on the development of metabolic dysfunction has not been reported yet (as far as we know). Most of the previous interventional clinical studies using HB-EGF modulators have focused on the treatment of cancer patients. 3,34,120,121
Antioxidant approaches against metabolic dysfunction
Obesity is closely associated with the induction of oxidative stress in adipose tissue and circulation. 51 There was a close association of oxidative stress with the development of metabolic syndrome. 52,84,118 As an example of the useful therapeutic application of antioxidants, the administration of a free thiol N-acetyl-L-cysteine ameliorated the liver endothelial cell damage caused by the paracetamol overdose. 122 –124 However, the beneficial effects of neutralization of the oxidants using antioxidants against metabolic disease phenotypes are still under controversy. 119,125 Because local transient ROS production is essential for physiological and cellular functions, 125,126 nonspecific targeting of superoxide (O2 −) and hydrogen peroxide (H2O2) may cause deleterious effects. Another pitfall of antioxidant therapy could be the differential reactivities of an antioxidant with oxidants due to different chemical structures and properties. 119 The time point for the administration might be another determining factor for therapeutic effects. 119,127 Administration of antioxidants at the end stage of metabolic diseases did not induce significant beneficial effects. 125,126
HB-EGF targeting against metabolic diseases
HB-EGF blocking antibodies were evaluated for therapeutic applications for antitumor purposes. 128 –132 In human clinical trials, the administration of HB-EGF neutralizing antibody showed protection against cancer progression. However, the administration induced unexpected psychiatric side effects. 130,133 A recent trial using another HB-EGF blocking antibody did not show the deleterious effects. 132 The membrane-tethered HB-EGF is the bona fide receptor for diphtheria toxin (DTX) in primates. 134 Inert DTX analog has been evaluated for the suppression of HB-EGF signaling in varying pathological conditions. 121,135 –137 A representative DTX analog, CRM-197, showed protection against the proliferation and metastasis of cancer cells 135 –139 ; however, the effects of the analog on the metabolic disease phenotypes have not yet been reported.
As mentioned, the administration HB-EGF ASO significantly downregulated the rate of hepatic VLDL production and effectively suppressed circulatory VLDL- and LDL-associated TG and cholesterol levels in hyperlipidemic mouse models (LDLR- or apoE-deficient mice under the Western diet). 92 Concordantly, the ASO administration induced an effective suppression of atherosclerosis and aneurysm developments in the models. 92,140 The inhibition of hepatic VLDL production and downregulation of innate immune cell production in the bone marrow appears to be involved in the protection. 14
Recently, a list of small-molecule EGFR blockers (e.g., gefitinib and AG1478) showed protection against atherosclerosis and hyperlipidemia in animal models. 102,141 The inhibitors also downregulated circulatory lipid levels in hyperlipidemic mouse models. The EGFR blocker erlotinib showed protection against diabetes in humans by suppressing inflammatory cytokine production and inflammatory cell infiltration into the pancreas. 142 Gefitinib administration protected against renal, vascular, and glomerular fibrosis. 109 Potentially, the re-evaluation of the clinically available EGFR blockers could be an approach for the development of therapeutic tools against metabolic diseases. 12,43
Conclusion and Perspectives
Since the first identification of the HB-EGF as a ligand of EGFR and ERBB4, 1 numerous features of HB-EGF associated with cell proliferation and metabolic disease phenotypes were reported. Different from the other EGFR ligands, the HB-EGF transcription is sensitively upregulated by oxidative stresses in the endothelial cells. The role of the HB-EGF in regulating the proliferation and differentiation of HSCs could be a compelling connection of oxidative stress with the induction of low-grade inflammation in obese people. The increase of the hepatic VLDL production by the HB-EGF signaling in the hepatocytes could be the linkage of oxidative stress with dyslipidemia in obese people. As illustrated in Fig. 3, the HB-EGF upregulated in the hepatic or bone marrow sinusoidal endothelial cells by overproduction of harmful oxidants in circulation appears to be a proximal event for the induction of dyslipidemia and low-grade inflammatory responses under oxidative stress environments. The vicious cycle of accumulation of oxidants in circulation and increased myeloid cell production in the bone marrow would lead to the progress of metabolic diseases to atherosclerosis and non-alcoholic steatohepatitis. HB-EGF is also involved in the induction of suppression of insulin signaling in the adipocytes and skeletal muscle cells.
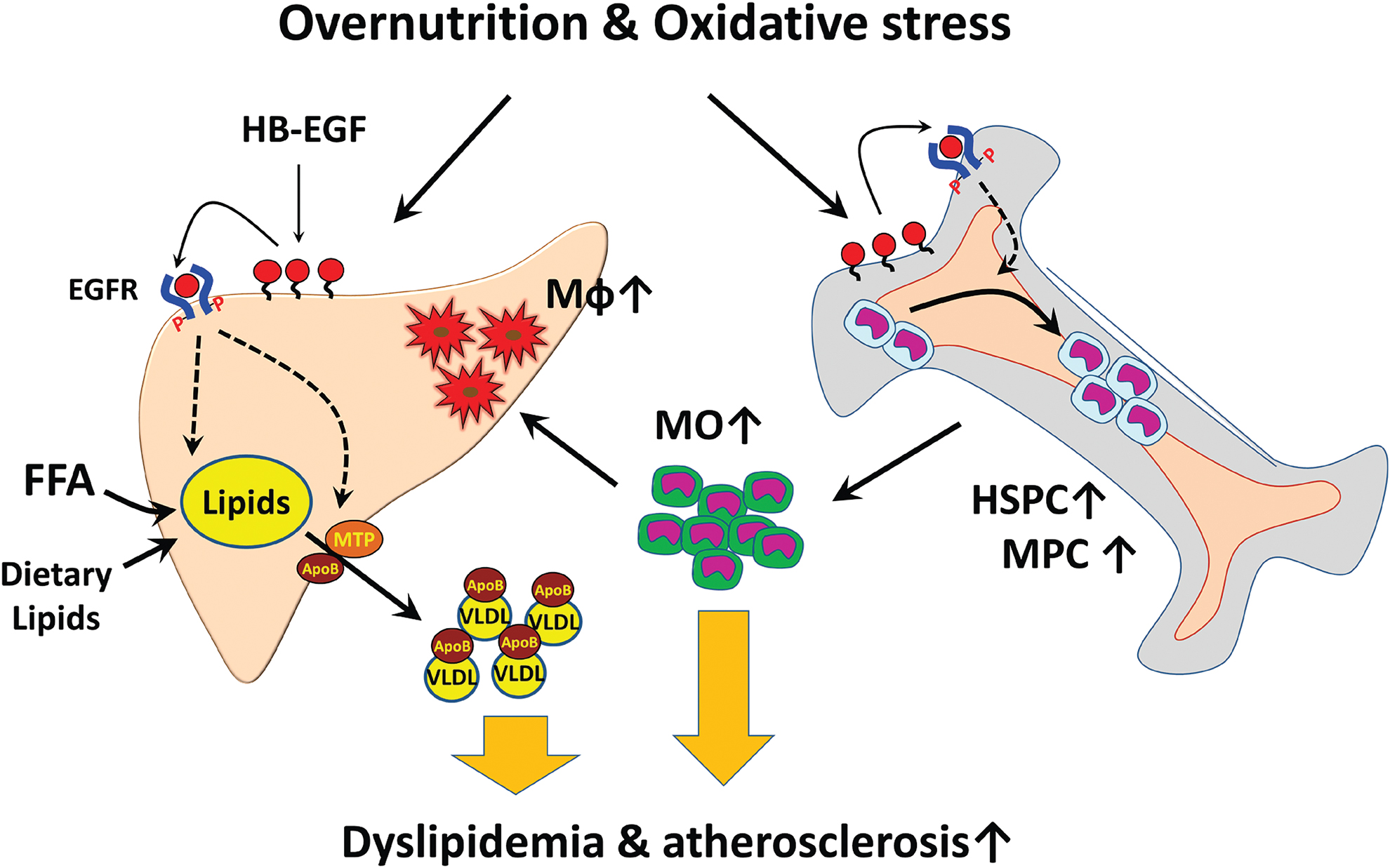
The working hypothesis of HB-EGF in metabolic disease development. Oxidants produced by the oxidative stress increased HB-EGF expression in the sinusoidal endothelial cells in the liver and bone marrow, causing increases in hepatic VLDL secretion and myeloid cell production. The bone marrow-derived monocytes can be recruited into the subendothelial space of the liver sinusoids and vessel endothelium for the development of inflammation in the liver and vessels. HB-EGF targeting would downregulate the hepatic VLDL production and reduction of the myeloid cell production. FFA, free fatty acid; HSPC, hematopoietic stem and progenitor cell; Mφ, macrophage; MO, monocyte; MPC, myeloid progenitor cell. Color images are available online.
Although the therapeutic potential of the HB-EGF modulation is a promising approach from the experimental results using animal models, still many barriers are to be overcome for the clinical applications. Previous reports indicated barriers in the translational application of the preclinical results to humans on metabolic and vascular disease phenotypes,
143,144
as exemplified by the increased susceptibility of the patients with anti-inflammatory reagent canakinumab (IL-1beta blocking antibody) administration to the sepsis and infection.
145
For each approach of HB-EGF targeting, a thorough evaluation of the potential side effects should be addressed. The administration of an HB-EGF blocking antibody caused psychiatric disturbances in human studies, partly because of the interruption of the cerebral function of HB-EGF.
130,133
The ASO would provide a benefit of minimal side effects in the brain via the impermeability of the ASO through the blood
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the views of fund supporters.
Authors' Contributions
S.K. made contributions to the acquisition and interpretation of experimental data associated with the article. V.S. and A.A.-L. provided insights into the experiments and direction of the projects related to the article. S.L. originally drafted the article with an agreement to be accountable for all aspects of the work.
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This study was supported by the National Institutes of Health K99/R00 HL105577 (S.L.) and the American Heart Association 17GRNT33700302 (S.L.).