
Abstract
The endoplasmic reticulum (ER), the center of protein folding, also controls the cell’s life-and-death signaling mechanisms. ER stress caused by unfolded or misfolded proteins leads to the activation of the unfolded protein response (UPR) in the cell. The UPR utilizes three main signaling pathways to restore disrupted ER homeostasis. These signaling pathways are protein kinase R-like endoplasmic reticulum kinase, inositol-requiring enzyme 1, and activating transcription factor 6. Studies have reported that ER stress (ERS) plays a role in the pathogenesis of metabolic disorders such as diabetes, obesity, atherosclerosis, and nonalcoholic liver disease. This review will briefly discuss the ERS response in these metabolic diseases.
Introduction
As an intracellular organelle, the endoplasmic reticulum (ER) plays a crucial role in protein biosynthesis and regulates lipid and calcium metabolism. 1 It is a significant intracellular calcium store and contributes to the biogenesis of autophagosomes and peroxisomes. 2 The ER carries a dense network of protein chaperones, prevents the backlog of unfolded or aggregated proteins, and regulates misfolded proteins trapped in low-energy kinetic traps. 3 Chaperones are a class of proteins that play a crucial role in cellular processes by assisting in protein folding, maintaining protein stability, and preventing protein aggregation. These molecular chaperones are essential for ensuring proper protein folding and preventing misfolding, which can lead to protein aggregation and cellular dysfunction. 4 Moreover, chaperones are involved in various cellular processes, including maintaining protein folding during ER import, chaperone-mediated degradation, and regulating protein quality control mechanisms. 5 These chaperone-mediated processes (the overabundant ER chaperone Binding immunoglobulin protein [BiP]/Glucose-regulated protein 78 [GRP78]) harness the power of metabolism to provide high-quality protein folding in the ER. 3 Putative chemical chaperones can inhibit ER stress (ERS) and reduce adipose tissue (AT) inflammation in obesity, thereby influencing changes in metabolic dysfunction. 6 Different disorders, such as nutrient deficiency, altered glycosylation at the cellular level, calcium dysregulation, oxidative damage, and posttranslational modification disorders, are referred to as ERS. As a result, unfolded proteins are stored in the ER lumen and can impair ER homeostasis. It can also activate a series of adaptive mechanisms known as unfolded protein response (UPR). 7 –9
The UPR has different effects depending on the degree of damage caused by ERS. With nominal ERS, normal cell function is restored. The UPR halts translational activities, inactivates signaling pathways that cause increased synthesis of extrafold proteins, and begins to address the problem of misfolded proteins. 10 The backlog of misfolded proteins and subsequent ERS is called proteotoxicity, which can contribute to developing diabetes, cancer, and neurodegenerative diseases. 9 Although the UPR attenuates ERS by degrading immature proteins in the ER, severe or extensive ERS leads to activation of the proapoptotic UPR and cell death. 11 In this review, we aimed to summarize the updated literature examining ERS and the metabolic diseases most affected by ERS. Figure 1 summarizes ERS and the associated metabolic diseases.
ER stress and associated metabolic diseases. ER, Endoplasmic Reticulum; PERK, Protein Kinase R-Like Endoplasmic Reticulum Kinase; ATF6, Activating Transcription Factor; XBP1, X-Box-Binding Protein 1; JNK, C-Jun N-Terminal Kinase, NAFLD; Nonalcoholic Fatty Liver Disease. (Created with BioRender.com).
The functions of the UPR include decreasing the amount of recently formed protein entering the ER lumen, increasing the protein folding capacity of the ER (through transcriptional upregulation of ER chaperones and folding catalysts), and finally stimulating the degradation of misfolded and aggregated protein. 12 Combinatorial signals from ER transmembrane detectors (IRE1, PERK, and ATF6) that initiate the UPR signaling pathway promote folding and maturation of secretory proteins by increasing transcription of objective genes encoding ER chaperones and enzymatic outputs. 13 X-box-binding protein 1 (XBP1), the essential UPR regulator, mediates ERS targeting. XBP1 spliced and XBP1 unspliced are two isoforms of XBP1. 14
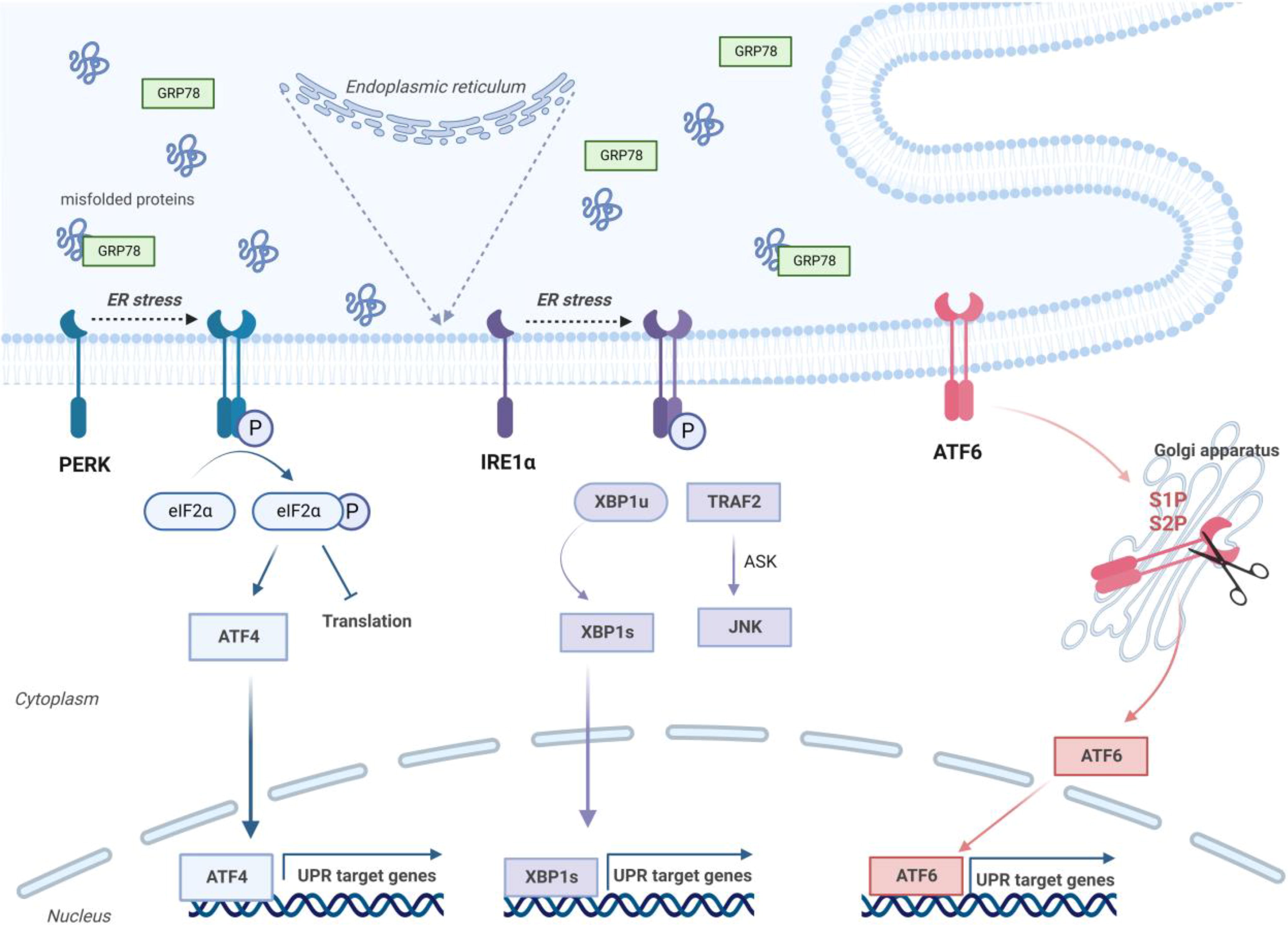
Inositol-requiring enzyme 1 (IRE1) is a type I transmembrane protein that implicates an ER-luminal domain for the identification of ERS signals and a cytosolic endoribonuclease domain for the catalysis of downstream processes, including the cleavage of XBP1u message RNA (mRNA) into XBP1s mRNA. XBP1s mRNA encodes the transcription factor XBP1s to transactivate many stress adaptation genes. 15
Protein kinase R-like endoplasmic reticulum kinase (PERK) is a type I transmembrane protein kinase localized to the ER membrane, and its C-terminal serine/threonine protein kinase domain is associated with the Eukaryotic initiation factor 2 alpha (eIF2α) kinase family. During ERS, GRP78 primarily dissociates from PERK, as unfolded or misfolded proteins in the ER competitively bind GRP78 and then cause disinhibition of PERK and mobilization of PERK through dimerization and autophosphorylation. 16
Activating transcription factor 6 (ATF6) exits the ER along the ERS and migrates to the Golgi to form a cytosolic fragment with transcription factor activity, where it is sequestered by the site1 protease and by the site2 protease. Several genes, including protein folding, lipid biogenesis, and ER-associated degradation, are stimulated by ATF6. In unicellular eukaryotes, the entire UPR is controlled by IRE1, whereas in metazoans, this control is exerted by PERK and ATF6 (Fig. 2). 17

The three main pathways (PERK, IRE1α, and ATF6) involved in the ER stress response. ER, Endoplasmic Reticulum; GRP78, 78 kDa glucose-regulated protein; PERK, protein kinase R-like endoplasmic reticulum kinase; ATF4, activating transcription factor 4; UPR: unfolded protein response; IRE1α, inositol-requiring enzyme 1α; XBP1, X-box-binding protein 1; TRAF2, Tumor necrosis factor (TNF) receptor associated factor-2 (TRAF2); ASK, apoptosis signal-regulating kinase; JNK, c-Jun N-terminal Kinase; ATF6, activating transcription factor. (Created with BioRender.com).
Activation of the UPR mediates the enlargement of the ER membrane and completes the expanded organelle gap with newly synthesized protein folding mechanisms, increasing the amount of ER as needed. 18 The UPR has been found to play an essential role in metabolism, inflammation, cell diversification, and survival. 19
UPR connection with inflammatory pathways
Both immune system cells and cells involved in metabolic pathways sense pathogens, irritants, and cellular damage, triggering the release of inflammatory agents, including cytokines, released radicals, hormones, and other small particles, and an inflammatory response begins. 20 ERS affects both the signaling pathways that lead to the production of inflammatory mediators and the reaction of cells to immunogenic stimuli. 21 Pathways triggered by the ERS response stimulate aseptic inflammation, and three detectors (IRE1, PERK, and ATF6) of the UPR pathway regulate the duration of inflammation. ERS plays an essential role in various cells in the development of several diseases, including obesity, type 2 diabetes, cancer, and intestinal and respiratory diseases; inflammation triggered by ERS is a significant contributor to disease progression. 22
Toll-like receptor (TLR) signaling stimulates endoplasmic reticulum stress (ERS) in macrophages, enhancing responses upon TLR binding to their ligands. Specifically, TLR2 and TLR4 trigger the activation of IRE1α and subsequently XBP1s, mediated by Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase NADPH oxidase 2 (NOX2) and tumor necrosis factor (TNF) receptor-associated 6. Knockdown of IRE1α in Murine macrophage cell line (J774) macrophage cells also reveals the significant impact of the IRE1α-XBP1s axis on TLR-mediated response by inhibiting the production of interleukin-6 (IL-6), TNF-α, a type of adipokine secreted by adipocytes, and interferon β. 23 ERS can also activate nuclear factor kappa B (NF-κB). 24 During impaired protein folding and binding in the ER, phosphorylation of eIF2α by Pancreatic endoplasmic reticulum kinase (PEK) (PERK or Eukaryotic translation initiation factor 2 alpha kinase 3 [EIF2AK3]) is necessary to stimulate NF-κB transcriptional activity. 25 Furthermore, ATF6 plays a role in activating NF-κB through its effect on the Mammalian target of rapamycin - protein kinase B (mTOR-AKT) signaling pathway. 24
Insulin resistance
Insulin resistance (IR) is the response of target tissues, such as fat and muscle tissue, especially the liver, to stimulation by insulin. In IR, impaired glucose excretion leads to increased insulin production and the development of hyperinsulinemia. It causes metabolic consequences such as hyperglycemia, increased inflammatory markers, hyperuricemia, dyslipidemia, visceral obesity, endothelial dysfunction, and hypertension. Advanced insulin resistance is also characterized by metabolic syndrome, nonalcoholic fatty liver disease (NAFLD), and type 2 Diabetes mellitus (DM). The etiology of insulin resistance can be acquired (which most often belongs to this group) or inherited, but it can also be both. 26
The main function of AT is to store the nutrients taken more than the body needs, such as triacylglycerol, and to release free fatty acids during starvation. Adipose tissue exerts its biological roles in an autocrine, paracrine, or systemic manner. In addition, it influences various physiological processes related to energy, glucose metabolism, and immunity and secretes more than 50 hormones and signaling molecules called adipokines. These adipokines also exhibit proinflammatory or anti-inflammatory properties and contribute to insulin resistance. In lean individuals, in the presence of metabolic disease, proinflammatory adipokines either directly affect the insulin signaling pathway or indirectly modulate insulin resistance via stimulation of inflammatory pathways. 27
Because insulin pro-receptors are proteolytically immature, ERS prevents the transport of recently synthesized insulin pro-receptors to the plasma membrane. 28 TNF-α increases insulin resistance by desensitizing insulin receptors. 29 Genetic manipulation of XBP1 in cultured cells and all animals results in causality between sensitivity to ERS and insulin resistance. Cells lacking XBP1 are significantly more susceptible to ERS and are subject to insulin resistance via IRE1α-dependent activation of c-Jun N-terminal kinase (JNK) and serine phosphorylation or damage to IRS1. In contrast, cells endowed with highly activated XBP1 resist ERS and protect themselves from insulin resistance. 30
Obesity
Food consumption is one of man’s greatest pleasures and, at the same time, an essential mechanism for survival. In modern society, overeating leads to obesity, which has become a global epidemic. 2 The World Health Organization (WHO) describes the global epidemic of overweight and obesity as an excessive or high backlog of body fat that poses a threat to health. The WHO describes normal weight, overweight, and obesity as a body mass index (BMI, calculated as weight in kg/height in m2) of 18.5–24.9, 25–29.9, and 30 or more, respectively. BMI categorizes obesity as grade I (30–34.9), grade II (35–39.9), and grade III or high obesity (≥40). According to the WHO BMI data, 43% of countries with current nutrition data report that half or more of the adult population has a BMI ≥ 25. 31 Obesity-induced inflammation may be described as low-grade, chronic inflammation because of surplus nutrients and energy. 32
Chronic ERS and dysfunction are a feature of obesity and thoughtfully contribute to meta-inflammation, aberrant hormonal activity, and altered substrate metabolism in metabolic structures such as liver and adipocytes. 33 Obesity induces ERS and inflammation in the liver by increasing blood glucose, free fatty acids (FFA), insulin, and proinflammatory cytokines, and also accelerates lipid accumulation by activating glycogen synthase kinase (GSK)-3β. 23,34
Various anti-inflammatory adipokines are secreted in the AT of lean individuals. These include adiponectin, transforming growth factor beta (TGFβ), interleukin (IL)-10, IL-4, IL-13, IL-1 receptor antagonist (IL-1Ra), and apelin. In contrast to lean individuals, obese AT secretes proinflammatory cytokines such as TNF-α, IL-6, leptin, visfatin, resistin, angiotensin II, and plasminogen activator inhibitor 1. 27
Obesity results in hypoxia as a consequence of inadequate AT oxygen supply. 35 Obesity is a chronic, low-grade form of inflammation because the AT contains large numbers of macrophages, which are a rich source of TNF-α and IL-6. 29 In a comparison between mice and lean animals fed incessantly with increased fat consumption, PERK and IRE1α phosphorylation and JNK activity were decreased in both fat cells and livers of high-fat-fed mice. 30
Diabetes mellitus
The main finding of diabetes mellitus is chronic hyperglycemia, and this term is commonly used for heterogeneous metabolic disorders. The reason for this may be the presence of impaired insulin secretion, impaired insulin action, or both of these. 36 There are two main types of diabetes: type I (10% of all people with diabetes) and type II (90% of all people with diabetes). 37 There is ample evidence that ERS may play a role in the pathogenesis and complications of diabetes. 38 For example, ERS can prevent STAT-3-dependent suppression of gluconeogenic enzymes and plays a vital role in increasing hepatic glucose production in obesity and diabetes. Because of the high insulin requirement, β-cells rely heavily on the ER to ensure proinsulin synthesis and appropriate folding. 39 Studies show that acute ERS arrested phosphorylation and nuclear input of transcriptional coactivator 2, which supported ATF6 expression and activated gluconeogenesis. 40
Attention to the UPR (specifically the PERK-eIF2α pathway) has been demonstrated by studies in PERK-deprived mice and in the context of Wolcott-Rallison syndrome, an uncommon childhood-onset insulin-dependent DM because of deprivation mutation in the PERK gene. 39 Thus, pancreatic β-cells lacking PERK are more sensitive to apoptosis stimulated by ERS, and mice lacking PERK ameliorate neonatal hyperglycemia caused by defective islet cell proliferation and increased apoptosis. 41 Furthermore, mice in which elF2α phosphorylation is disrupted ameliorate diabetes because of irregulated proinsulin translation, increased oxidative damage, decreased expression of β-cell-related genes, and apoptosis. 42 Following ER stress, UPR activation attempts to restore the disrupted ER balance. If the balance is not restored, proapoptotic and proinflammatory signaling pathways are activated. C/EBP homologous protein (CHOP), NF-κB, GRP78, and XBP1 are activated via the ATF6 signaling pathway, and the cell is primed for apoptosis. 37
Atherosclerosis
Atherosclerosis, which is related to ischemic diseases such as myocardial infarction and cerebral infarction, is mainly caused by metabolic disorders. 43 Protein quality control is significant for efficient cardiomyocyte contraction. Impaired protein homeostasis leads to cellular dysfunction and cell death. 23 Studies have shown three different ways ER function can be effective in the progression of atherosclerosis. First, a direct link between lipid metabolism and UPR has been demonstrated. For example, XBP1 has been shown to play a role in synthesizing phosphatidylcholine in the ER and expanding the ER membrane. Studies in the literature show that ERS contributes positively to lipogenesis and hepatic lipid deposition, but there needs to be more information on the specific activity of signaling pathways in the UPR. The chemical or molecular solubility of ERS prevents hepatic lipid accumulation. It facilitates lipoprotein secretion while increasing inhibition of phospholipid synthesis or phospholipase activity, leading to changes in phospholipid metabolism and exacerbating ERS and sphingolipid levels, which can affect the current function of the ER. 2. During ERS, hyperglycemia, hepatic glucose production stimulation, and glucose release suppression occur because ERS causes abnormal insulin action. It is known that hyperglycemia, hyperinsulinemia, or both can bridge the gap between obesity, type 2 DM, and atherosclerosis.
Because ERS triggers free cholesterol-induced cell death in macrophages, the ER can sense stress related to lipid status and transmit this knowledge to pathways involved in inflammation and death, at least to some types of cells that are important for disease progression. The ERS-activated UPR plays a role in the adaptive immune system, mainly by reducing the continuum and presentation of peptides associated with the major histocompatibility complex (MHC-1) induced by excess saturated fat. 44 The UPR is activated in endothelial cells (EC) in response to the lesion and various metabolic and physical effects. A high expression level of XBP1 in developed plaque areas, especially in ECs, increases the violence of atherosclerosis. In cultured human aortic ECs, tunicamycin-induced ERS can activate the production of essential cytokines such as IL-6, IL-8, and monocyte chemoattractant protein-1, which can be prevented by silencing XBP1 or ATF4. Oxidized 1-palmitoyl-2-arachidonyl-sec-3-glycerophosphorylcholine, homocysteine, and hydrolyzed low-density lipoprotein (LDL), in which oxidized and glycated LDL induce UPR signaling pathways, may contribute to the progression of atherosclerosis through the induction of ERS and inflammatory pathways. 24
Nonalcoholic fatty liver disease
NAFLD is often considered a hepatic manifestation of metabolic syndrome (IR, obesity, and hyperlipidemia), including hepatic steatosis (NASH). NASH is characterized by lipid accumulation, hepatocyte death, inflammation, and fibrosis. It carries the risk of liver cirrhosis, hepatocellular carcinoma, and liver-related mortality.
45
The data show that the relationship between fatty acids and NASH is also essential. Saturated fatty acids, which include palmitate and stearate but not their monounsaturated conjugates, stimulate ERS and contribute to the development of obesity, metabolic syndrome, and cardiovascular disease. Saturated fatty acids are pro-inflammatory, while unsaturated omega-3 fatty acids, such as docosahexaenoic acid and eicosapentaenoic acid, have opposite effect.
46
Fatty acids are physiologically essential
Studies show that ERS contributes to the amelioration of NAFLD by affecting hepatic lipid metabolism by activating lipogenesis and inhibiting very low-density lipoproteins. Studies are showing decreased expression of Apolipoprotein B100 (ApoB100), an increased lipogenesis gene expression associated with decreased Phosphorylated eukaryotic initiation factor 2 alpha (p-eIF2a) in the livers of CD154 (CD40LG) knockout mice fed olive oil.
49
The ER requires a high concentration of Ca
Conclusion
ER, which is involved in the biosynthesis of lipid and carbohydrate conjugates, is also involved in protein folding, assembly, and biosynthetic transport. ER, which is involved in the biosynthesis of lipid and carbohydrate conjugates, is also involved in protein folding, assembly, and biosynthetic transport. 52 Different intracellular and extracellular stresses can increase the number of misfolded proteins, leading to an imbalance in ER homeostasis known as ERS. 53 ERS occurs when the metabolic function of the endoplasmic reticulum is impaired as a consequence of the imbalance between energy intake and expenditure. ERS has been correlated with metabolic syndromes like IR, obesity, DM, and NAFLD. ERS occurs when the endoplasmic reticulum’s metabolic function is impaired because of the imbalance between energy uptake and consumption. ERS has been linked to metabolic syndromes such as IR, obesity, DM, and NAFLD. 54 Once we fully understand the impact of the UPR pathway on metabolic diseases and discover the possible mechanisms, we can explore potential treatment areas and prepare efficient intervention strategies.
Footnotes
Authors’ Contributions
Fi.S. and H.V.: Idea and concept—constructing the hypothesis or idea of research and/or article. F.S.: Design—planning methodology to reach the conclusions. H.V.: Control and supervision—organizing, supervising the course of progress and taking the responsibility of the research/study. Fi.S. and F.S.: Data collection and processing—taking responsibility in patient follow-up, collection of relevant biological materials, data management and reporting, execution of the experiments. Fi. S and F.S.: Analysis and interpretation—taking responsibility in logical interpretation and conclusion of the results. Fi. S and F.S.: Literature review—taking responsibility in necessary literature review for the study. F.S.: Writing—taking responsibility in the writing of the whole or important parts of the study. H.V.: Critical review—reviewing the article before submission scientifically besides spelling and grammar. H.V.: References and funding—providing personnel, environment, financial support tools that are vital for the study. H.V.: Materials—biological materials, taking responsibility of the referred patients.
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
No funding was received for this article.