
Abstract
Evidence is presented that components of fetal calf serum (FCS) can significantly enhance the splicing correction activity of peptide nucleic acids (PNA) in HeLa pLuc 705 cells. The effect proved more pronounced for PNAs bearing fluorescence tags and relies on the ability of specific components of FCS to mediate a mainly nonendocytotic intracellular delivery of PNA. Attempts to isolate and characterize the active serum components using PNA-loaded beads and nano-LC-ESI mass spectrometry revealed the growth-factor related inter-alpha-trypsin inhibitor and the adhesion protein fibronectin to be substantially responsible for the delivery activity of FCS.
Introduction
In line with the latter reports, previously we found evidence for an extensive ability of PNA to cross mammalian plasma membranes (Oehlke et al., 2011) and to exert antisense activity in primary heart cells (Oehlke et al., 2004; Turner et al., 2010). Our results implied cell-type-dependent export by protection machineries rather than poor penetration into cells to be responsible for the mostly weak intracellular activity of PNA (Oehlke et al., 2011). On the other hand, naked PNA in our hands also showed no detectable activity in the splicing correction assay developed by Kole et al. (Kang et al., 1998; Wolf et al., 2006).
In the current study, we have repeated the splicing correction experiments under modified conditions, assuming the association propensity of PNA and the presence of serum to be most important for the ambiguity of our previous results. Indeed, in contrast to our previous study, we found significant splicing correction activity for naked PNA at the low micromolar level if the incubation was performed in the presence of fetal calf serum (FCS). Moreover, the activity could be enhanced further by more than one order of magnitude by tagging the PNA with a fluorescence label and heating the PNA solution before addition of serum. Our results suggest specific components of FCS to associate with PNA and to mediate a mainly nonendocytotic cellular uptake of PNA. First attempts to identify such serum components by means of a PNA-loaded resin and liquid chromatography-tandem mass spectrometry point to the growth-factor related inter-alpha-trypsin inhibitor and the adhesion protein fibronectin to be main constituents of the vector activity contained in FCS. The proteins found appear promising as potential physiological delivery vehicles for PNA without the limitations on toxicity and immune response of the currently used viral and chemical vectors.
Materials and Methods
General
Chemicals and reagents were purchased from Sigma or Bachem unless specified otherwise. The reference 2′-O-methyl-oligonucleotide (5′-CCU CUU ACC UCA GUU ACA-3′) was synthesized by Eurogentec S.A.
Synthesis of PNA
PNA oligomers were manually synthesized using the t-Boc strategy (Christensen et al., 1995) The peptide segments of the conjugates were prepared by the solid-phase method using standard Boc chemistry (Stewart and Young, 1984). To introduce the fluorescent label, the deprotected N-terminus of the PNAs or the peptides were reacted in dimethylformamide (DMF) for 3 days at room temperature with 10 equivalents of 5(6)-carboxyfluorescein-N-hydroxysuccinimide ester (FLUOS; Boehringer) or for 1 hour with 3 equivalents of dansyl chloride or NBD-chloride in the presence of 6 equivalents of diisopropylethylamine. Purification was carried out by semipreparative HPLC on Vydac C18 using a 250×8 mm column. MALDI-MS (Voyager-DE STR BioSpectrometry Workstation MALDI-TOF; Perseptive Biosystems, Inc.) provided the expected [M+H]+ peaks.
Cell culture
HeLa pLuc 705 cells were cultured in DMEM containing 4.5 g/L glucose (Biochrom) supplemented with 10% FCS (Biochrom) and 1% (v/v) nonessential amino acids at 37°C in a humidified 5% CO2 containing air environment.
CHO (Ham's F12), MDCK (DMEM), or HEK (DMEM) cells were analogously grown.
Splicing correction assay
For assessing the splicing correction according to Kole et al. (Kang et al., 1998), normally HeLa pLuc 705 cells were plated in 96-well plates at a density of 105 cells per well and cultured overnight. The culture medium was discarded, and the cells were washed twice with PBS. The cells were incubated for 24 hours at 37°C with the PNA in 200 μL/well DMEM containing 10% FCS in a humidified 5% CO2 containing air environment. After 24 hours, the cells were washed twice with icecold PBS and overlaid with 30 μL/well of each DMEM and ONE-GLo™ Luciferase Reagent (Promega). After lysis for 5 minutes at ambient temperature, 50 μL/well of the resulting solutions were used for measuring the luminescence by a safire2 microplate reader (Tecan) and each 3–5 μL for quantifying the cellular protein according to Bradford (1976).
In the cases of a PNA-treatment for only 4 hours, the cells were subsequently washed twice with PBS and cultured for another 20 hours in 200 μL/well DMEM containing 10% FCS as described earlier. This procedure was used also for substituting FCS by 1% bovine serum albumin (BSA), 0.1% BSA and endothelial cell growth factor (EGF) (Sigma; Prod. No. E9640) or 0.1% BSA and fibronectin bovine (Sigma; Prod. No. F1141).
If the incubation was performed at a low temperature, then the cells were cooled in the original medium for 30 minutes at 0°C, then washed twice with 0°C PBS, and incubated for 2 hours with the PNA in 200 μL/well DMEM containing 10% FCS at 0°C. Incubation was continued without replacing the incubation solution for another 22 hours at 37°C in a humidified 5% CO2 containing air environment.
Heating the PNA solution before the incubation was performed for 60 minutes at 60°C after having dissolved the PNA at the ten-fold of the required concentration in DMEM. Subsequently, one part of this solution was immediately mixed with 9 parts of 37°C DMEM containing 10% FCS and added within 15 minutes to the cells.
FACS analysis
Cells were plated in 6-well plates at a density of 5×105 cells per well and cultured overnight as described earlier (see Cell culture section). After washing thrice with Dulbecco's phosphate-buffered saline containing 1 g/L D-glucose (DPBSG), the cells were overlaid with 1 mL of a freshly prepared prewarmed (37°C) 5 μM solution of Fl-PNA-1 in DMEM without or with 9% FCS, respectively. After 60 minutes of incubation at 37°C, the cells were washed with 0°C DPBS and detached by 5 minutes of treatment at 0°C with 1 mL of 0.1% Pronase E (Sigma) in DPBS containing 0.5 mM EDTA. Subsequently, 700 μL of PBS containing 1% BSA were added, the cell suspension was centrifuged at 1000 g for 4 minutes at 4°C, and the pellet was resuspended in 1 mL of 0.1% BSA/PBS. The accumulation of fluorescence was determined at 525 nm with excitation at 488 nm using a Becton Dickinson FACS Calibur flow cytometer with CELLQUEST software. Cytograms were acquired with 104 cells.
Isolation and characterization of PNA-binding FCS components
To 1.3 mL of 10% FCS/H2O were given 1 mg of Tenta Gel S RAM resin (Rapp Polymere GmbH) in untreated form or covalently loaded with 0.23 mmol/g PNA-1, respectively. The suspensions were gently shaken for 60 minutes at ambient temperature. Subsequently, the resins were spun down and washed 4 times with each 100 μL of H2O. Then, the resins were heated in each 50 μL of sample buffer Roti-Load 1 (Carl Roth GmbH) for 5 minutes at 95°C, and each 30 μL of the resulting extracts were subjected to SDS-PAGE (10% acrylamide). After separation, the gel was stained with Coomassie Imperial Protein Stain (Thermo Fisher Scientific), and 2 mm slices were excised. Tryptic digest of proteins and nanoLC-MS/MS experiments were performed as previously described (Lange et al., 2010). Briefly, tryptic peptides were separated by a reversed-phase capillary liquid chromatography system (Eksigent 2D nanoflow LC; Axel Semrau GmbH) connected to an LTQ-Orbitrap XL mass spectrometer (Thermo Scientific). Mass spectra were acquired in a data-dependent mode with one MS survey scan (with a resolution of 60,000) in the Orbitrap and MS/MS scans of the 5 most intense precursor ions in the LTQ. The MS survey range was m/z 350–1500. The dynamic exclusion time (for precursor ions) was set to 120 sec, and automatic gain control was set to 3×106 and 20,000 for Orbitrap-MS and LTQ-MS/MS scans, respectively. For database searching, raw data files were processed with the Mascot Distiller (version 2.2.1; Matrix Science) to generate Mascot generic files (mgf files). The generated peak lists (mgf) and the MASCOT server (version 2.2; Matrix Science) were used to search in-house against the SwissProt 2010_7 protein database (517100 sequences; 182146551 residues). A maximum of 2 missed cleavages was allowed, and the mass tolerance of precursor and sequence ions was set to 10 ppm and 0.35 Da, respectively. Acrylamide modification of cysteine and methionine oxidation were considered as possible modifications. Scaffold (version 2.1.03; Proteome Software, Inc.) was used to validate MS/MS based peptide and protein identifications and to generate nonredundant protein lists. Peptide identifications were accepted if they could be established at greater than 70.0% probability, as specified by the Peptide Prophet algorithm (Keller et al., 2002). Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. Based on decoy database searches, the false positive rate was estimated to be <1%.
Results and Discussion
In previous studies, we have found evidence that export processes counteracting the cellular uptake rather than inability to cross plasma membranes should be responsible for the often observed poor intracellular activity of naked PNA (Turner et al., 2010; Oehlke et al., 2011). In contrast to these findings, however, we failed in line with numerous reports of others to detect splicing correction activity for a naked 18mer PNA targeted to the cryptic splice site of luciferase pre-mRNA (PNA-1, Table 1) (Kang et al., 1998; Wolf et al., 2006). To obtain more detailed information about the reasons for this failure, in the current study, we have tested the activity of the previously used PNA sequence in the splicing correction assay according to Kole et al. (Kang et al., 1998) under modified conditions.
PNA, peptide nucleic acids.
Splicing correction activity of naked PNA under conditions counteracting the export of internalized PNA
In our previous study, we have incubated cells with PNA according to the mostly used protocol for a few hours in the absence of FCS, followed by a further culturing over night without PNA in the presence of FCS (Wolf et al., 2006). Within the latter period, however, the concentration gradient caused by the absence of extracellular PNA could favor a possible export of internalized PNA. To obtain information whether such export can be counteracted by maintaining the external PNA concentration during the entire culturing period, we have exposed cells to the PNA in the presence of FCS for 24 hours. Under these conditions, we found a moderate but statistically significant splicing correction activity of naked PNA at concentrations higher than 5 μM (Fig. 1). In principle, this finding supports the reports claiming the ability of naked PNA to exert significant biological activity in mammalian cells also in the absence of delivery vectors (Oehlke et al., 2004; Turner et al., 2010). A 10 μm solution of scrambled PNA of identical base composition (PNA-2, Table) did not show any effect (not shown) implicating the observed splicing correction activity to be sequence specific (it was found to be true also after the below-described foregoing heating and also for Fluos- and PNA-2; Table 1). Initial incubation at 0°C did not reduce the splicing correction activity, thereby indicating an at least partial nonendocytotic mode for the cellular uptake of the PNA (Fig. 1). On the other hand, the intracellular activity found indicates an only very weak capability of naked PNA to enter the cell interior. The intracellular activity was found to be more than 2 orders of magnitude lower than that achieved in the same cell batch with a 2′-O-methyl-oligonucleotide ([5′-CCU CUU ACC UCA GUU ACA-3′]; 1 μM; 330 relative luminescence units [RLU]/μg protein) complexed with lipofectamine (50 μg/mL), being the common reference.
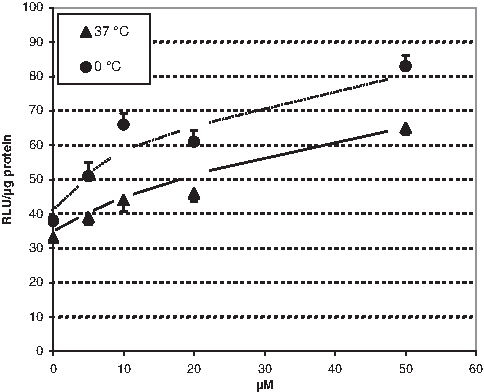
Concentration dependence of splicing correction according to Kole et al. (Kang et al., 1998). Luminescence intensities measured in the cell lysates after exposing HeLa pLuc 705 cells to different concentrations of PNA-1 for 24 hours at 37°C or for 2 hours at 0°C followed by 22 hours at 37°C. Incubation in the same manner with 10 μM scrambled PNA-2 remained without effect (36±3.7 and 38±1.6 RLU/μg protein at 37°C and 0°C, respectively). Data are expressed in relative luminescence units (RLU)/μg protein. Each point represents the mean of 6 samples±SEM. The difference between the controls and the 5 μM values are statistically significant at P≤0.02 (Student's t-test).
This only limited intracelluar availability of naked PNA should normally be too low for saturating the often micromolar target levels in transfected cells but may be sufficient to saturate the natural low nanomolar ones in primary cells. In this context, these results possibly resolve the ambiguity between, on one hand, reports that are claiming significant intracellular activity of naked PNA and, on the other hand, such denying it.
Incubation in the presence of FCS combined with a foregoing heating resulted in a significantly enhanced splicing correction activity of various PNA derivatives
Heating of the PNA solution at 60°C immediately before adding FCS significantly enhanced the intracellular PNA activity (Fig. 2), thus indicating monomeric PNA formed by thermal decomposition of hydrogen bonded associates to be the active PNA entities. Heating procedure proved even more effective for fluorescence labeled PNA (Fig. 2).

Splicing correction of various naked PNAs after heating and subsequent incubation in the presence of FCS. Luminescence intensities measured after exposing HeLa pLuc 705 cells to each 5 μM of PNA-1 and its fluorescence-labeled analogs for 24 hours at 37°C without and with heating the PNA solutions immediately before the addition of FCS for 60 minutes at 60°C (FLUOS-, 5(6)-carboxyfluorescenyl; NBD-, 7-nitrobenz-2-oxa-1,3-diazol-4-yl; DNS-, 3-(5-dimethylaminonaphthalene-1-sulfonyl). Data are expressed in RLU/μg protein. Each bar represents the mean of 6 samples±SEM. The differences between the control (no PNA) and the values of the not heated samples as well as between the not heated and the respective heated samples are statistically significant at P<0.02 (Student's t-test).
Statistically significant splicing correction results have been achieved under these modified conditions even after only 4 hours incubation for 5 μM PNA (Fig. 3), thus making comparative studies possible. When the incubation with PNA was performed in the absence of FCS or rather in the presence of 1% BSA, the heating effect disappeared nearly completely (Fig. 3). These observations indicate that the thermal decomposition of PNA associates alone is not sufficient for enhancing the splicing correction activity and suggest a complementing contribution of FCS. That this complementing effect might rely on a simple stabilization of decomposed PNA entities by nonspecific adsorption to serum proteins is rendered unlikely by the failure of BSA to substitute the FCS effect. A peculiarity of the used serum batch could be ruled out by testing 3 other FCS batches which led to almost identical results (not shown). Taken together, these observations suggest interactions between specific FCS components and PNA to be responsible for the found enhanced splicing correction activity. Likewise, imply the strong effects of heating that the putative FCS components are unable to interact with hydrogen-bonded PNA-associates formed in the stock solutions and require their thermal decomposition to monomeric entities for mediating the intracellular PNA activity.
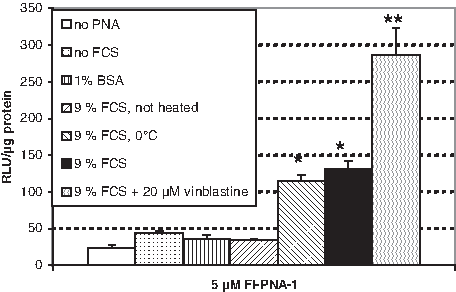
Splicing correction of Fl-PNA-1 after incubation under various conditions. Luminescence intensities measured after exposing HeLa pLuc 705 cells for 4 hours to 5 μM of Fl-PNA-1 after foregoing heating for 60 minutes at 60°C, in the presence and absence of FCS, in the presence of 1% bovine serum albumin (BSA), and in the presence of both FCS and 20 μM vinblastine at 37°C or in the presence of FCS at 0°C (in this case, the cells were precooled for 30 minutes at 0°C before the incubation), followed by washing twice with PBS, and further 20 hours of culturing in DMEM/10% FCS. Data are expressed in RLU/μg protein. Each bar represents the mean of 6 samples±SEM. The differences between the nonlabeled and the asterisk-labeled bars as well as between the single- and double-asterisk-labeled bars are statistically significant at P<0.02 (Student's t-test).
To obtain information whether the effects of the putative FCS components may rely on an enhanced cellular uptake or only on an effect on the intracellular splicing correction process, we have performed FACS analysis after incubating HeLa pLuc 705 cells either in the presence or in the absence of FCS with and without foregoing heating of the PNA solution. We found a clearly higher fluorescence intensity for cells treated with heated PNA in the presence of FCS than without heating or in the absence of FCS, respectively (Fig. 4). This pattern goes well along with the splicing correction activities observed under the respective conditions (Fig. 3) and suggests these activities to be at least partially related to the cellular uptake of PNA. An analogous pattern was found also after FACS analysis of various cell types (Table 3) and after incubation with an unrelated 15-mer PNA (Fl-GAG CCA CAG AGG TAG-Lys-NH2) using the same conditions (not shown). These findings confirm the effects of heating and FCS to be independent on peculiarities of the HeLa pLuc 705 cells or of sequence or chain length of the PNA and suggest a more general delivery ability of the involved FCS components.
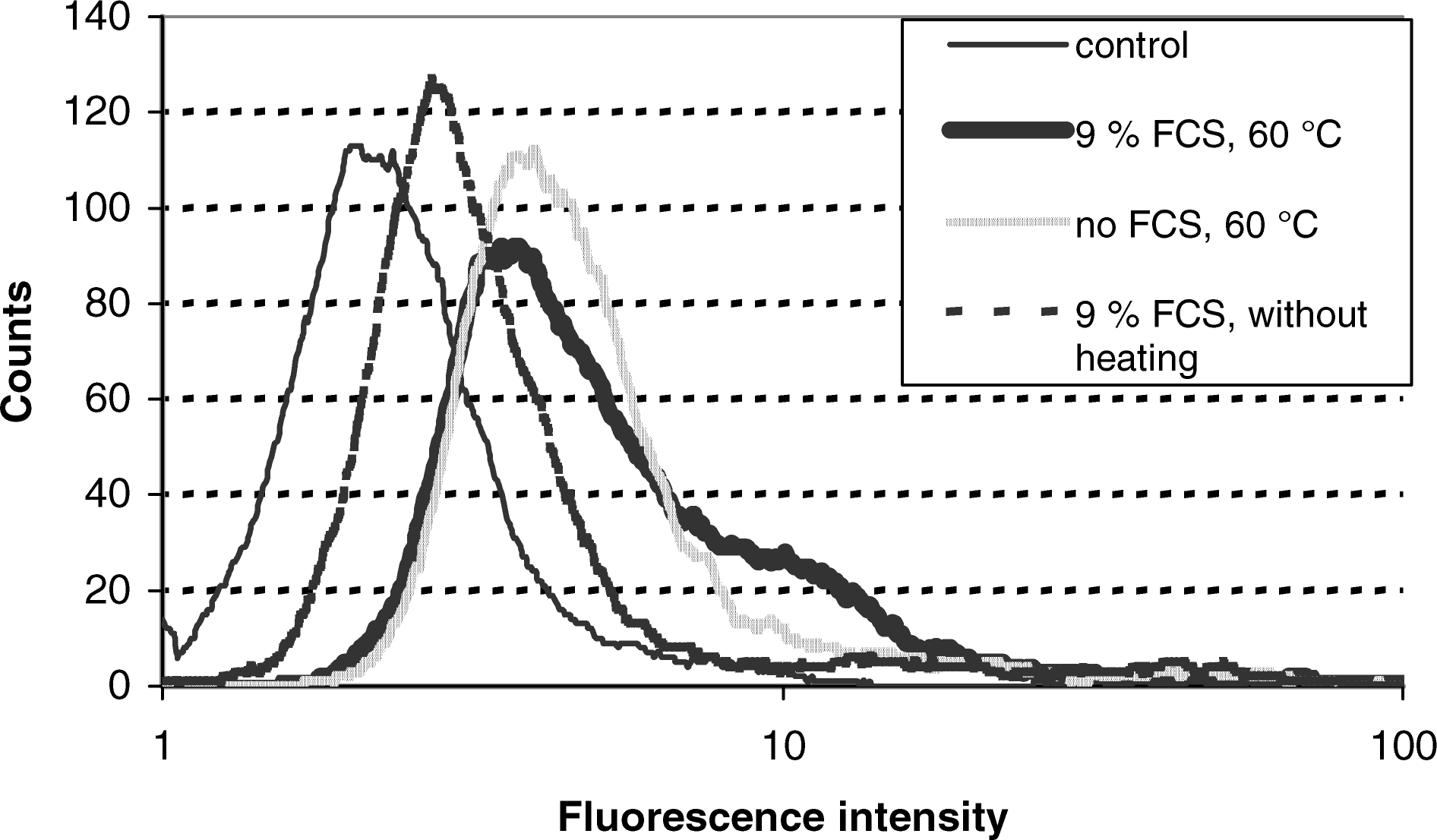
FACS analysis of HeLa pLuc 705 cells after incubation with Fl-PNA-1 under various conditions. Cell-associated fluorescence measured by flow cytometry after exposure of HeLa pLuc 705 cells for 60 minutes at 37°C to empty buffer or to 5 μM Fl-PNA-1 heated or not heated in advance for 60 minutes at 60°C in the presence or absence of 9% FCS, respectively. (Thin line: empty buffer, mean fluorescence intensity 3.1; bold line: PNA heated, 9% FCS, mean fluorescence intensity 7.3; broken line: PNA not heated, 9% FCS, mean fluorescence intensity 5.7; gray line: PNA heated, without FCS, mean fluorescence intensity 6.5). Cell number is plotted on the ordinate as a function of the fluorescence intensity on the abscissa.
Confocal laser scanning microscopy experiments in HeLa pLuc 705 cells loaded with the differently labeled naked PNAs did not reveal significant differences with regard to both location and intensity (normalized to that of the respective incubation solutions) of the intracellular fluorescence (not shown). Thus, the respective activity differences displayed in Fig. 2 should be related to yet nonidentified factors, like, for example, the affinity to the target, rather than to different intracellular availabilities.
The putative FCS components mediate an at least partial nonendocytotic cellular uptake of PNA
The presence of 100 μM chloroquine, known as an endosome disrupting reagent (Midoux et al., 1993; Erbacher et al., 1996), during the entire 24 hours culturing period had no effect on the splicing correction activity of PNA-1 and Fl-PNA-1 (not shown). This finding indicates the PNA activity to be not primarily dependent on an escape from endosomes and, therefore, infers a mainly nonendocytotic mode of the cellular uptake of PNA. This notion is supported by the observation that incubation at 0°C did only moderately reduce the PNA activity (Fig. 3).
Exposing the cells to the PNA in the presence of vinblastine, a reagent inhibiting ABC-transporters such as P-glycoprotein (Cole et al., 1992; Gottesman and Pastan, 1993), led to a more than 2-fold increased activity (Fig. 3), thereby suggesting the cellular uptake of PNA under normal conditions to be counteracted by vinblastine sensitive export pumps. This finding is in line with results of our previous studies that suggest the intracellular availability of PNA to be substantially influenced by energy dependent transport machineries (Turner et al., 2010; Oehlke et al., 2011).
Isolation and characterization of PNA-binding FCS components
The known ability of transcription factor-like proteins to associate with nucleic acids and to pass the cell membrane in a nonendocytotic way (KUCHLER, 1993) tempted us to speculate that the here-observed FCS mediated cellular uptake of PNA might rely on an association with transcription factor-related serum components. To find support for this hypothesis, we have performed fishing experiments using PNA-loaded- (PNA-1, Fl-PNA-1, PNA-2) and unloaded synthesis resin (see Isolation and characterization of PNA-binding FCS components section). These exploratory experiments revealed, in line with our assumption, moderate delivery activity for the DMEM extracts of the FCS-treated PNA-loaded synthesis resins, but no effect of those of the unloaded resin (not shown). Therefore, we have separated the fished proteins by SDS-PAGE and identified them by nano-LC-ESI tandem mass spectrometry after in-gel digestion with trypsin (see Isolation and characterization of PNA-binding FCS components section). Foregoing exploratory dialysis experiments had indicated at least 90% of the delivery activity of FCS to be contained in the serum fraction with a molecular weight higher than 100 kDa (not shown), so that the mass spectrometric protein identifications were focused on this range. By this procedure 2 proteins have been detected in the extracts of the PNA-loaded resin in an amount which clearly exceeded that found in the extract of the unloaded resin (Table 2). Identification by means of the SwissProt 2010_7 protein database revealed these proteins to be inter-alpha-trypsin inhibitor and the adhesion protein fibronectin (Table 2). The first of the 2 appeared well reconcilable with the anticipated hypothesis if regarding inter-alpha-trypsin inhibitor as a precursor of the EGF 2b (Gebhard et al., 1988). The second hit, the adhesion protein fibronectin, on the other hand, seemed to be without importance in this context.
The statistical significance of all respective differences between the mean fluorescence intensities of cells treated in the absence or in the presence of FCS (each 10,000 measured cells) corresponds to levels of P<0.01 in the Student's t-test.
Nevertheless, testing the commercially available EGF in place of inter-alpha-trypsin inhibitor and fibronectin in a concomitant administration with PNA to cells revealed clear effects of both on the splicing correction activity (Fig. 5). The effects reached a maximum at a concentration of around 30 nM for both proteins (molecular masses: EGF 18 kDa, fibronectin 272 kDa), suggesting saturation for the involved transport machineries at this concentration range. In the absence of PNA or in the presence of its scrambled analog PNA-2, the splicing correction activity of cells treated with both proteins (0.77 or 7.7 μm/mL EGF or fibronectin, respectively) proved virtually identical with that of untreated cells (not shown), thus indicating side-effects of these proteins alone unlikely to play a considerable role here.
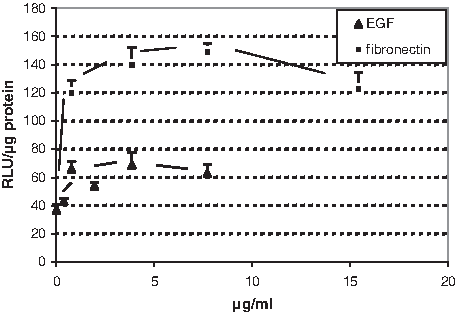
Splicing correction of Fl-PNA-1 after incubation in the presence of various concentrations of endothelial cell growth factor (EGF) or fibronectin. Luminescence intensities measured after exposing HeLa pLuc 705 cells for 4 hours to 5 μM of Fl-PNA-1, heated before for 60 minutes at 60°C, in the presence of various amounts of EGF or fibronectin dissolved in DMEM containing 0.1% BSA, followed by washing twice with PBS and further 20 hours of culturing in DMEM/10% FCS. Data are expressed in RLU/μg protein. Each bar represents the mean of 6 samples±SEM.
Incubation at lower temperature or in the presence of chloroquine or vinblastine revealed only moderate effects on the delivery activity of both EGF and fibronectin (Fig. 6), thus suggesting nonendocytotic transport processes to be involved. Similar results found in the presence of FCS makes it likely that EGF or its parent protein inter-alpha-trypsin inhibitor and fibronectin indeed belong to the putative PNA vectors contained in FCS.

Splicing correction of Fl-PNA-1 after incubation under various conditions in the presence of EGF or fibronectin. Luminescence intensities measured after exposing HeLa pLuc 705 cells for 4 hours to 5 μM of Fl-PNA-1, heated before for 60 minutes at 60°C, in the presence of 0.77 μg/mL EGF
Ongoing studies using alternative protocols, cells, and sera are conducted to identify further vector components and to elucidate their potential for physiological delivery vehicles of PNAs and probably other bioactive molecules.
Conclusions
The intracellular activity of PNA, in particular after N-terminal tagging with aromatic residues, may be significantly enhanced by incubation in the presence of serum and a heating of the PNA solution before addition of FCS. The results are indicative of specific components in FCS that have the potential to deliver PNA extensively in a nonendocytotic way across mammalian plasma membranes. Evidence was found that EGF and the adhesion protein fibronectin belong to these potential physiological delivery vehicles for PNA contained in FCS.
Footnotes
Acknowledgments
The authors thank G. Vogelreiter, J. Eichhorst, A. Klose, D. Krause, H. Nikolenko, and H. Stephanowitz for excellent technical assistance.
Author Disclosure Statement
No competing financial interests exist.