
Abstract
Epigenetic modifications of N-terminal histone tails, especially histone H3, are important for the regulation of the target genes in chromatin. Specific methods for detection of these modifications in histone H3 N-terminal peptides are valuable tools for diagnostic and therapeutic purposes. As an alternative to antibodies, RNA aptamers display compatible binding affinities and selectivites against various biologically relevant targets. Systematic evolution of ligands by exponential enrichment (SELEX) was performed against histone H3R8Me2sym. A 14-amino acid peptide that mimics this modified histone tail was prepared in a biotinylated form and 10 selection cycles of SELEX were carried out. This produced 4 aptamers, one of which (clone 1) was observed to have low nanomolar binding affinity (Kd = 12 nM) against the cognate peptide. The affinity of this aptamer is comparable to 2 commercially available antibodies against differently modified histone H3 peptides and it displays a greater selectivity than the antibodies.
Introduction
Phosphorylation, methylation, and acetylation are the most frequent chemical modifications involved in PTMs in histones. All these epigenetic changes modulate chromatin states that eventually control the relevent gene expression. PTMs cause gene expression patterns that give rise to all typical cancer characteristics (ESTELLER, 2008; Rodríguez-Paredes and Esteller, 2011). Therefore, the detection of PTM of histone is meaningful to provide a tool for early cancer detection. These epigenetic modifications are typically detected using specific antibodies derived from the cognate epigenetic modifications (Hirota et al., 2005; Tjeertes et al., 2009). However, owing to the cross-activity of antibodies, agents that more specifically detect PTMs are required to differentiate more sophisticated modifications. Further, as novel modifications are being continuously discovered, an even greater need exists to develop systematic tools for the detection of PTMs in histone.
Among the 4 histone proteins, the N-terminal H3 is the most frequently modified by phosphorylation, acetylation, and methylation. Phosphorylation and acetylation are relatively easy to detect, because these processes cause reasonably large changes in the size, shape, and electrochemical properties of histones. As a result, a wide variety of antibodies can be used to detect these types of modifications with high levels of selectivity (Tapia et al., 2006, and references therein). In contrast to phosphorylation and acetylation, methylation induces relatively small changes in the basicities and hydrophobicities of histones (Smith and Denu, 2009). In addition, diverse modes of methylation on Arg can occur including monomethylation as well as 2 kinds of dimethylation (symmetric and asymmetric) (Bedford et al., 2007). As a consequence of difficulties associated with differentiation of Arg methylations, few antibodies have been developed to detect this type of PTM.
Our recent interest has focused on determining how additional modifications are made when the N-terminus of histone H3 contains preexisting modifications. For example, as phosphorylation at S10 by aurora B kinase is one of the most important histone modifications (Han et al., 2011), it is important to know how this process is influenced by preexisting modifications. During the course of the studies aimed at this goal, we observed that symmetric dimethylation at R8 leads to total abolishment of phosphorylation at S10 (Han et al., 2011). Consequently, we believed that the development of tools to elucidate the mechanism(s) of regulation of phosphorylation at S10 by dimethylation at R8 would be valuable, as inhibitors of aurora B kinase represent an interesting approach to anticancer chemotherapy (Lens et al., 2010).
RNAs are potentially important biopolymers for target recognition, because they are readily available and they possess unique conformations and molecular diversity. As a result, the RNA aptamer technology has been widely used for generating antibody-like molecules against many biologically relevant targets (Yan and Levy, 2009). As N-terminal histone proteins have extensively varied sequences, especially in histone H3 and H4 (Peterson and Laniel, 2004), the development of aptamers against these targets is a worthy goal. RNA aptamers should possess advantages over antibodies because of the short time periods that are required for their generation and selection and the fact that they are limited by the sizes of the targets. Importantly, although an RNA aptamer has been found to be selective against the acetylated histone H4 peptide (Lin et al., 2009; Williams et al., 2009), none that selectively recognize methylated Arg in the H3 histone peptide currently exists. In the effort described later, we have discovered an aptamer that is selected against the N-terminal H3 histone protein containing a symmetrical dimethyl modification at Arg. The selected aptamer was found to display nanomolar binding against the modified peptide as well as a selectivity that is superior to those of commercially available antibodies.
Materials and Methods
Syntheses of peptides
All peptides were prepared using a standard solid phase peptide synthesis method. Fmoc-protected biotinylated Lys was used to produce C-terminal biotinylated peptides. The synthethized peptides were purified using HPLC with a Zorbax C18 (3.5 μm, 4.6 × 150 mm) column and a gradient comprised of 5 minutes with 5% B followed by a linear gradient of 5%–70% B over 35 minutes [buffer A: water with 0.1% (v/v) trifluoroacetic acid (TFA); buffer B: acetonitrile with 0.1% (v/v) TFA]. The identities of the peptides were confirmed using mass spectrometry (MS) (VoyagerTM MALDI-TOF; Applied Biosystems), which gave the following results: 14-amino acid (aa) H3R8Me2sym biotin: MS [M+H]+: 1743.0 (calcd.), 1743.0 (obsd.); 14-aa nonmodified H3 biotin: MS [M+H]+: 1715.0 (calcd.), 1715.0 (obsd.); 14-aa H3K9Me2 biotin: MS [M+H]+: 1743.0 (calcd.), 1743.0 (obsd.). Chromatograms of peptides are shown in Fig. 1.
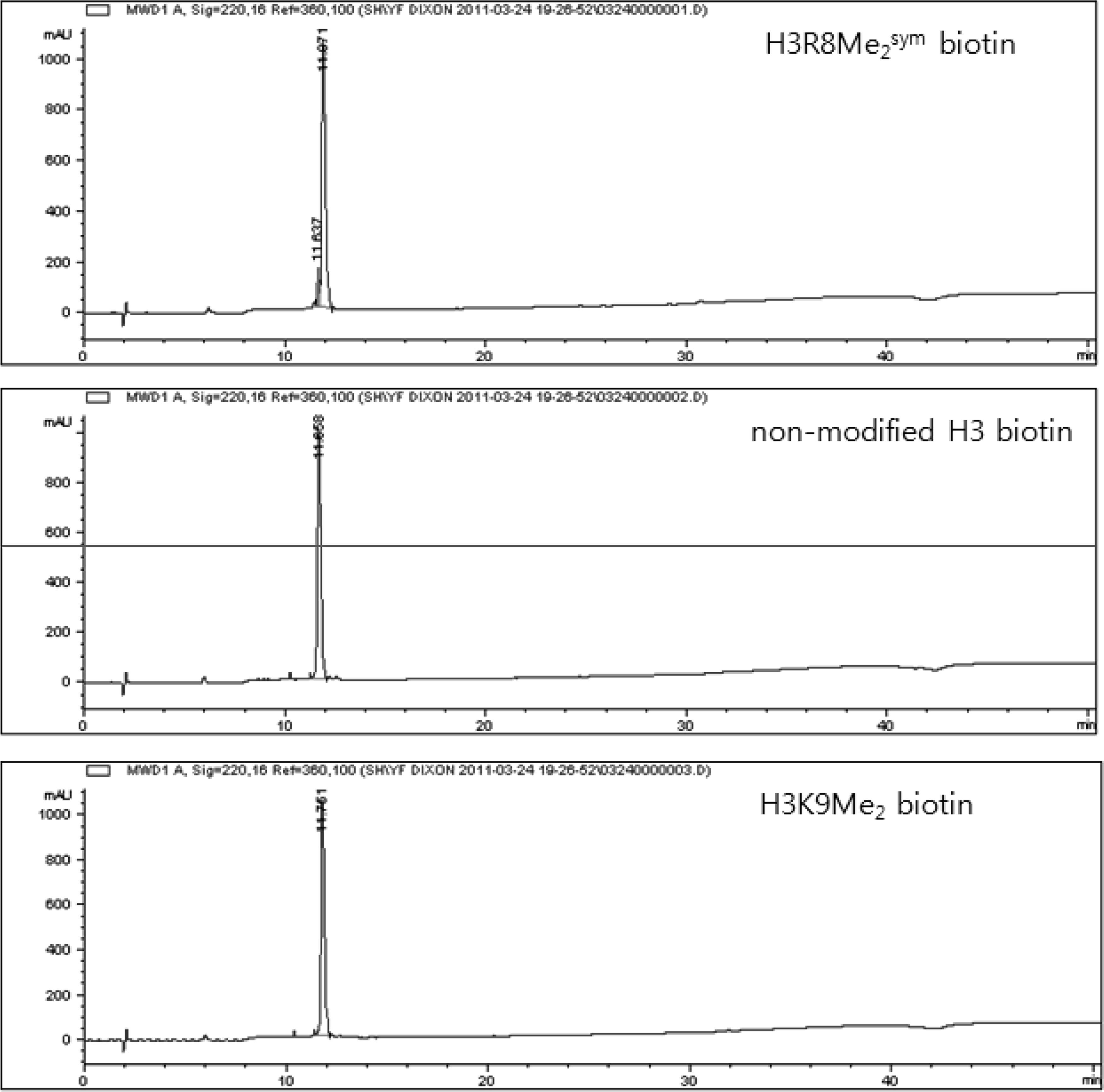
Chromatograms of purified peptides by HPLC A C18 column and gradient aqueous acetonitrile (5–70%) were used as the stationary and mobile phases respectively.
Procedures for SELEX cloning and sequencing of the selected RNA
The procedures for systematic evolution of ligands by exponential enrichment (SELEX) and cloning were carried out in the manner previously reported (Lee et al., 2009).
Binding affinity measurement for aptamers
BIAcore 3000 was used for the surface plasmon resonance (SPR) technique. The standard amine coupling method was used to immobilize streptavidin. Briefly, the carboxymethyl dextran matrix of a CM5 sensor chip was activated by injection of 70 μL of a coupling solution [100 mM N-ethyl-N'-(dimethylaminopropyl)carbodiimide and 25 mM N-hydroxysuccinimide]. The streptavidin solution (0.05 mg/mL) in 10 mM NaOAc (pH 5.5) was then injected into the activated flow cell. Streptavidin was immobilized ca. 4500 RU in each flow cell. The unreacted NHS esters were deactivated by injection of 70 μL of 1 M ethanolamine (pH 8.5). Flow cell 1 was biotinylated and used as a reference. Biotinylated H3R8Me2sym, nonmodified H3, and H3K9Me2 were injected into the respective streptavidin-coated flow cells 2–4. The final change in RU of each H3 peptide was ca. 600. For the binding assay, the flow rate was adjusted to 30 μL/minute. Various concentrations of the selected aptamer (clone 1, 2, 13, and 29) were injected using the serial automated method, which is comprised of sample injection (60 μL), dissociation (240 seconds), and regeneration (10 μL of 20 mM NaCl and 0.2 mM NaOH). The signals from a reference flow cell were subtracted from the observed signals (Figs. 2 and 3). Binding affinities of the aptamers and antibodies to the H3 14-aa peptide were calculated using BIAevaluation software (Tables 2–4 using a 1:1 Langmuir binding model). Anti-H3K9Me2 antibody and anti-H3 antibody were purchased from Upstate (no. 07-212) and Abcam (no. ab18521), respectively.
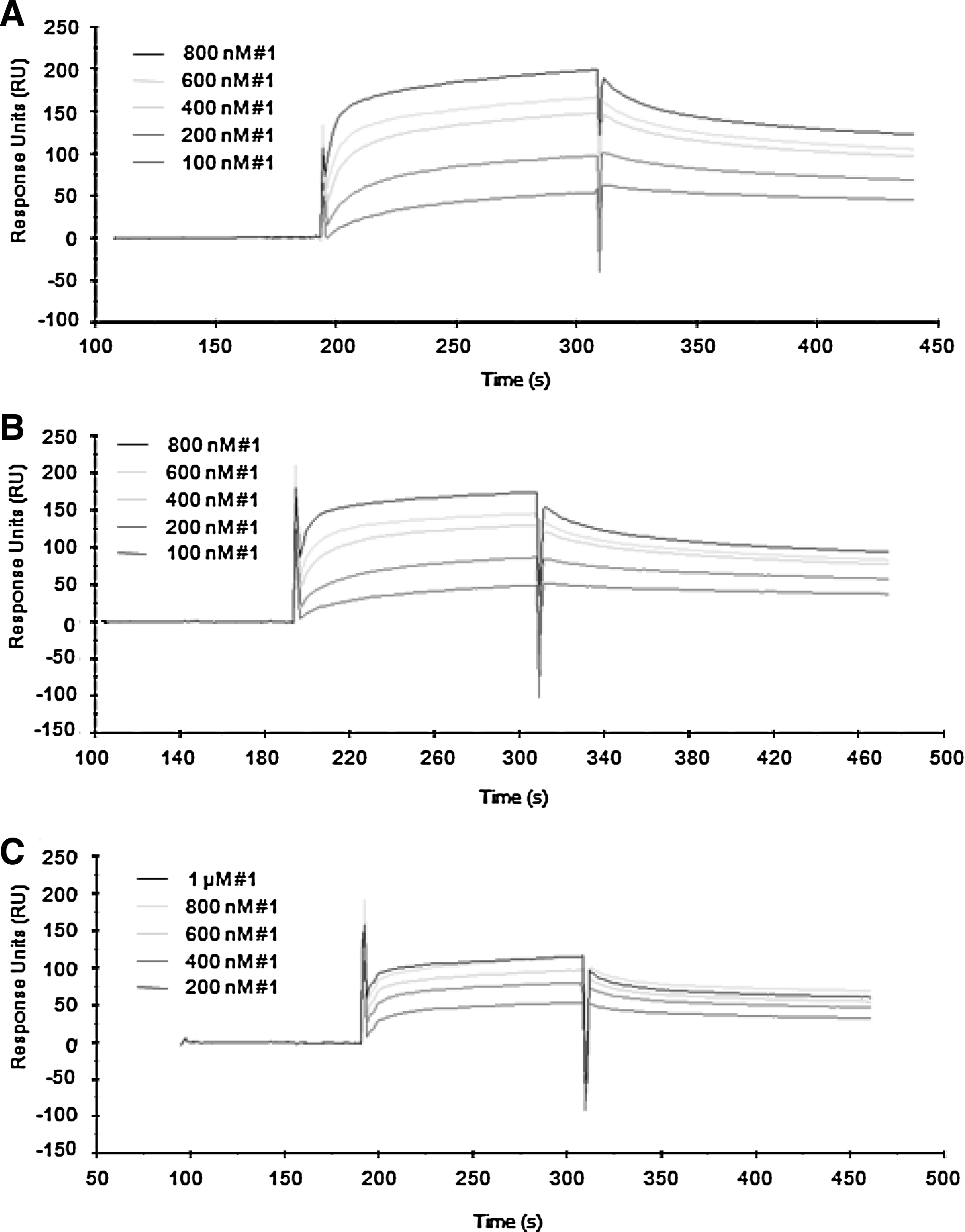
Binding affinity measurements of clone 1 aptamer by using the SPR technique.
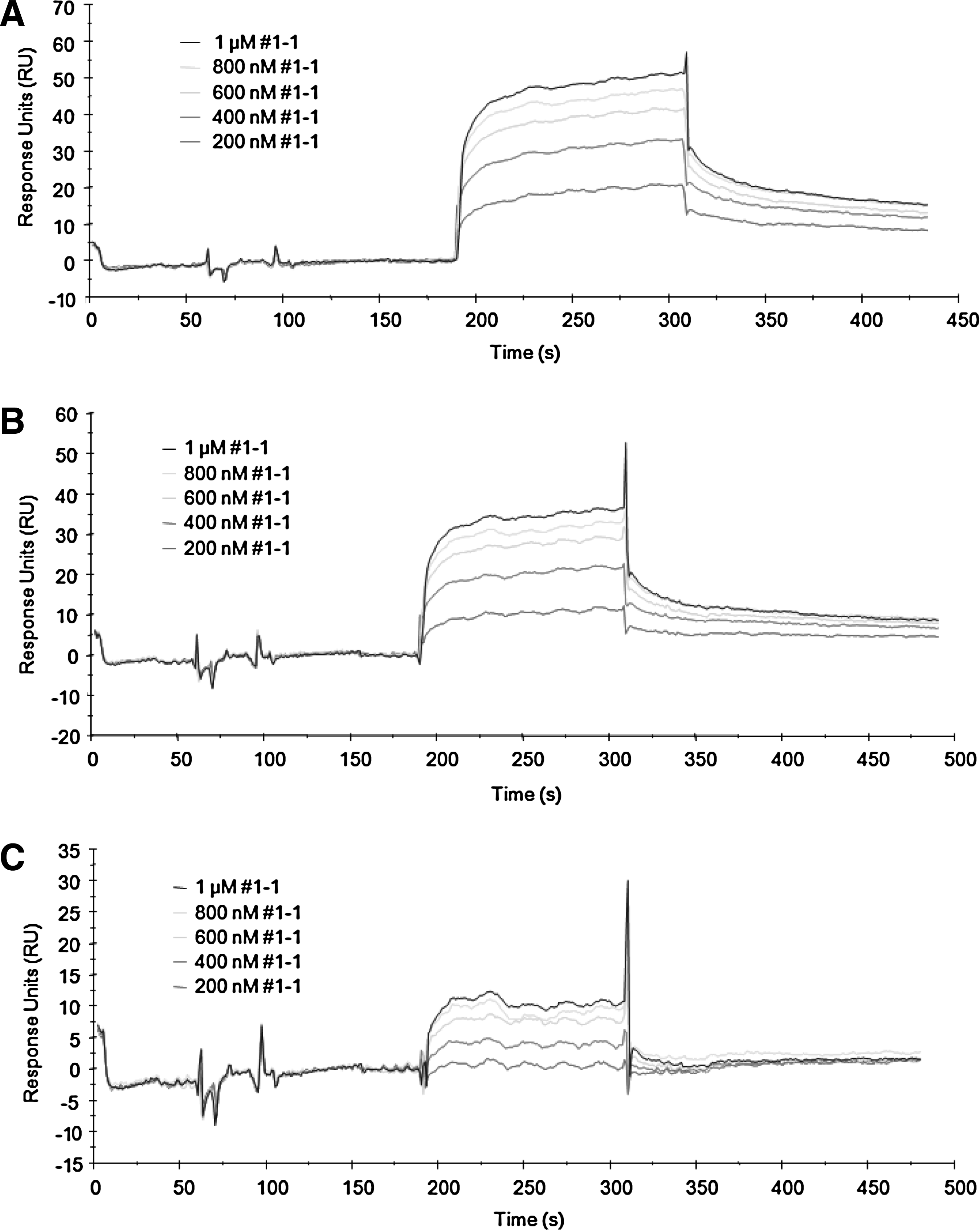
Binding affinity measurements of truncated hairpin 1-1 by using SPR technique.
Results and Discussion
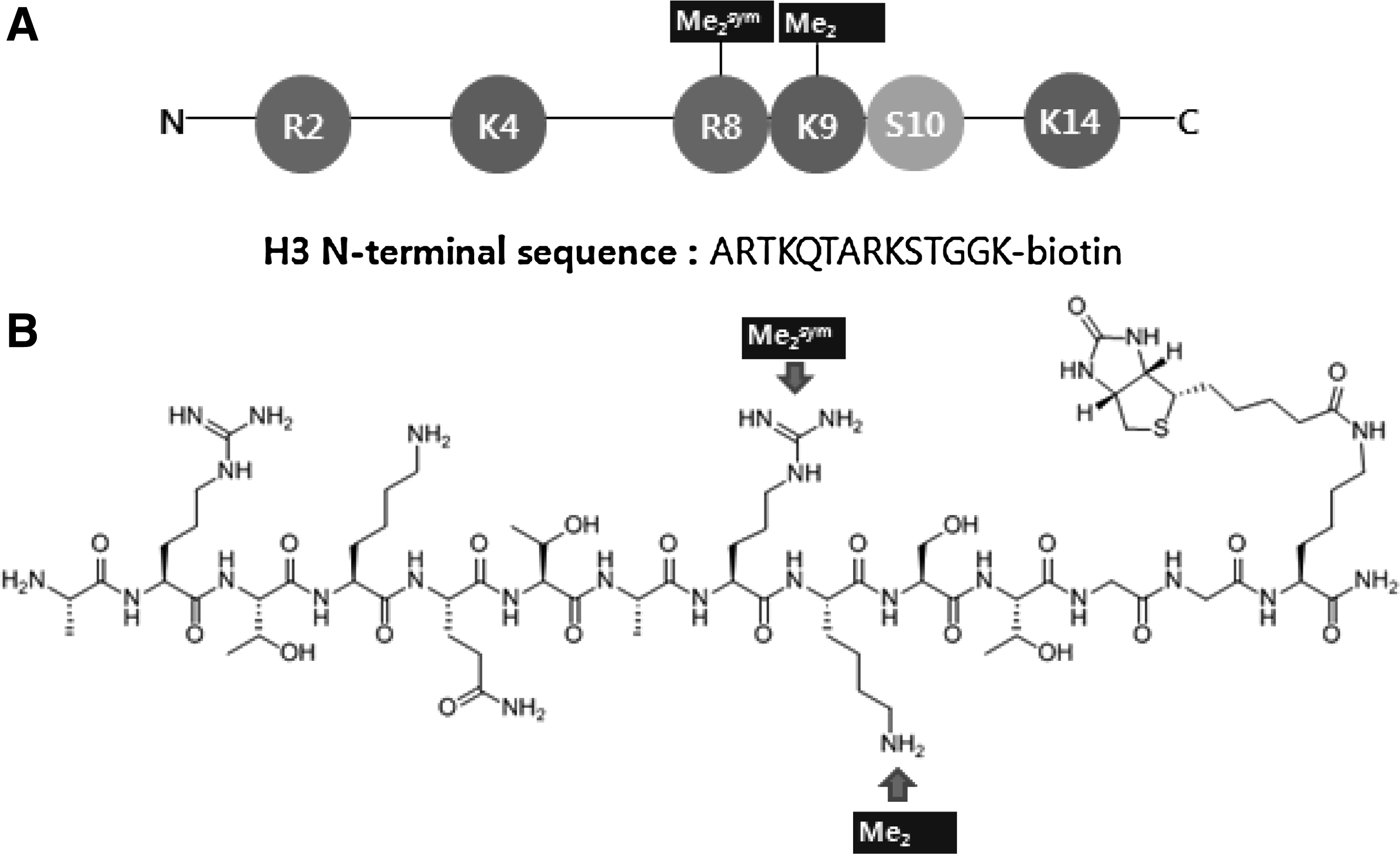
At the beginning of this effort, an attempt was made to synthesize the 16-aa peptide ARTKQTARKSTGGKAP, which has exactly the same sequence as is present at the end of N-terminal histone H3 with symmetrical dimethylation at R8 (H3R8Me2sym). Unfortunately, this approach was not successful. As an alternative, the 14-aa peptide (ARTKQTARKSTGGK) was successfully synthesized using standard Fmoc chemistry. To facilitate aptamer selection, the C-terminus of the peptide was biotinylated and the N-terminus was not acetylated to mimic the natural peptide sequence (Fig. 4). As references, 14-aa peptides matching the end of the N-terminal histone H3 but not containing modifications and dimethylation at K9 (H3K9Me2) were also prepared. The peptides were purified using HPLC and their structures were confirmed using MALDI-TOF mass spectrometry.
SELEX was carried out to produce aptamers against the 14-aa N-terminal H3 peptide containing the R8Me2sym modification. An RNA library containing 50 random nucleotides was designed and used for the cycles of SELEX. The selection procedure was carried out in the manner previously described (Lee et al., 2009). During the course of 10 SELEX cycles, the intensities of the DNA bands increased, indicating that the enrichment of eluted RNA was increasing (Fig. 5). After 10 cycles of selection, a pull-down assay showed that H3R8Me2sym-specific RNA aptamers from the normal RNA library had been enriched. The band intensities of eluted 10-cycled RNA pools against H3R8Me2sym and the control linker were 21% and 11%, respectively, under the given experimental conditions. Enriched aptamers from the RNA library were cloned to afford 4 different sequences (Table 1). One multiple clone and a few single clones were sequenced. Using SPR technique, the binding affinities of the 4 cloned aptamers were determined and all shown to be in the nanomolar range against modified as well as unmodified peptides (Fig. 2 and Table 2). Even though the library was enriched and converged to clone 2, one of the single cloned aptamers (clone 1) was found to display the highest binding affinity (Kd = 12 nM). Clone 1 not only had the strongest binding affinity against the cognate H3R8Me2sym peptide, but it also displayed the highest binding specificity for H3R8Me2sym in contrast to other modified and nonmodified peptides. The clone 29 aptamer also had a preferred selectivity against the cognate peptide, but other clones (including clone 2) displayed no selectivity. Interestingly, clone 2 and 13 aptamers showed selectivity for the H3K9Me2 peptide and the nonmodified peptide, respectively, indicating that each recognizes different sites of the peptide fragment. Thus, clones 2 and 13 are also plausible to be further studied as aptamers against nonmodified H3 histone tail. We, however, decided to focus on the H3K9Me2 aptamer in this moment, because the well-studied antibodies are already commercially available. As a consequence of the observations that clone 1 displayed a high selectivity and affinity for the cognate peptide, further studies focused on this aptamer. The clone 1 aptamer consists of 2 major hairpins based on an M-fold secondary structure prediction (Fig. 6A). Consequently, we prepared 2 individual truncated hairpins (hairpins 1-1 and 1-2 shown as C and D, respectively, in Fig. 6). Based on a pseudo-knot prediction, another structure is possible for clone 1 (Fig. 6B). As a result, a truncated hairpin RNA possessing a pseudo-knot structure (hairpin 1-3 in Fig. 6E) was also prepared. Binding affinities of the 3 truncated hairpins (hairpins 1-1, 1-2, and 1-3) against the peptides were determined using the SPR technique (Table 3). Among the 3 truncated hairpin RNAs, hairpin 1-1 had the strongest binding affinity (Kd = 71 nM) against the cognate peptide. However, binding is ca. 5 times weaker relative to the binding affinity of the whole clone 1 aptamer. In addition, the selectivity ratio of binding by hairpin 1-1 (1.8–3.5) is only one half of that of the whole clone 1 (3.5–7.0). The comparable affinities of hairpin 1-2 and 1-3 against the peptides suggest that 2 grooves from each of these hairpins cooperate, although not equally, in forming the peptide binding site. The observation of reduced selectivity in all 3 truncated hairpins of the clone 1 aptamer also supports the cooperative binding proposal. Even though it has the largest length among the 3 hairpins, the pseudo-knot structured 1-3 hairpin did not display a large binding affinity against the peptides. This finding indicates that the contribution of the pseudo-knot structure is not dominant and is perhaps negligible.
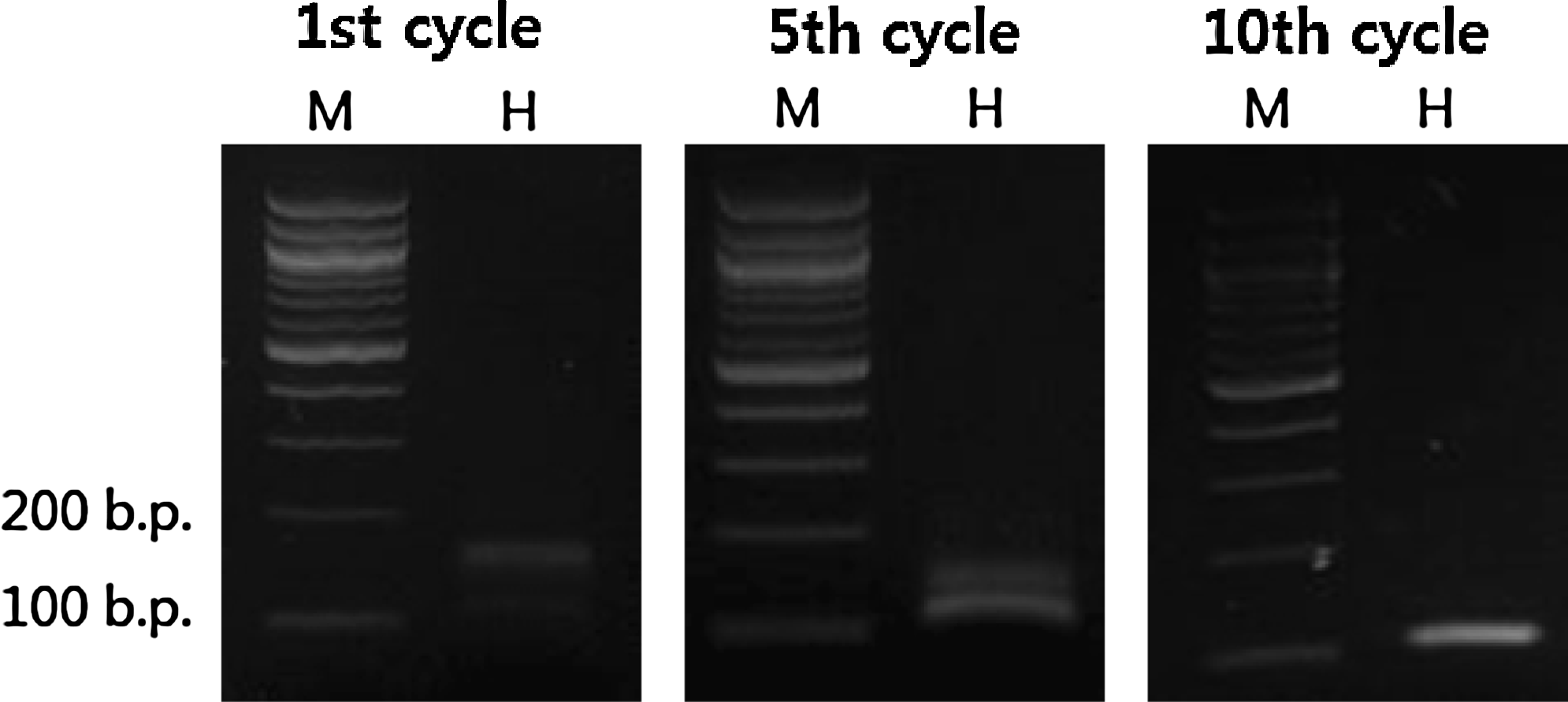
Aptamer selection process. Same amounts of the PCR mixtures were loaded at each selection cycle. The intensities of DNA bands increased compared with that of a DNA marker. M, 100-bp marker; H, PCR product for H3R8 Me2sym-systematic evolution of ligands by exponential enrichment; PCR, polymerase chain reaction.
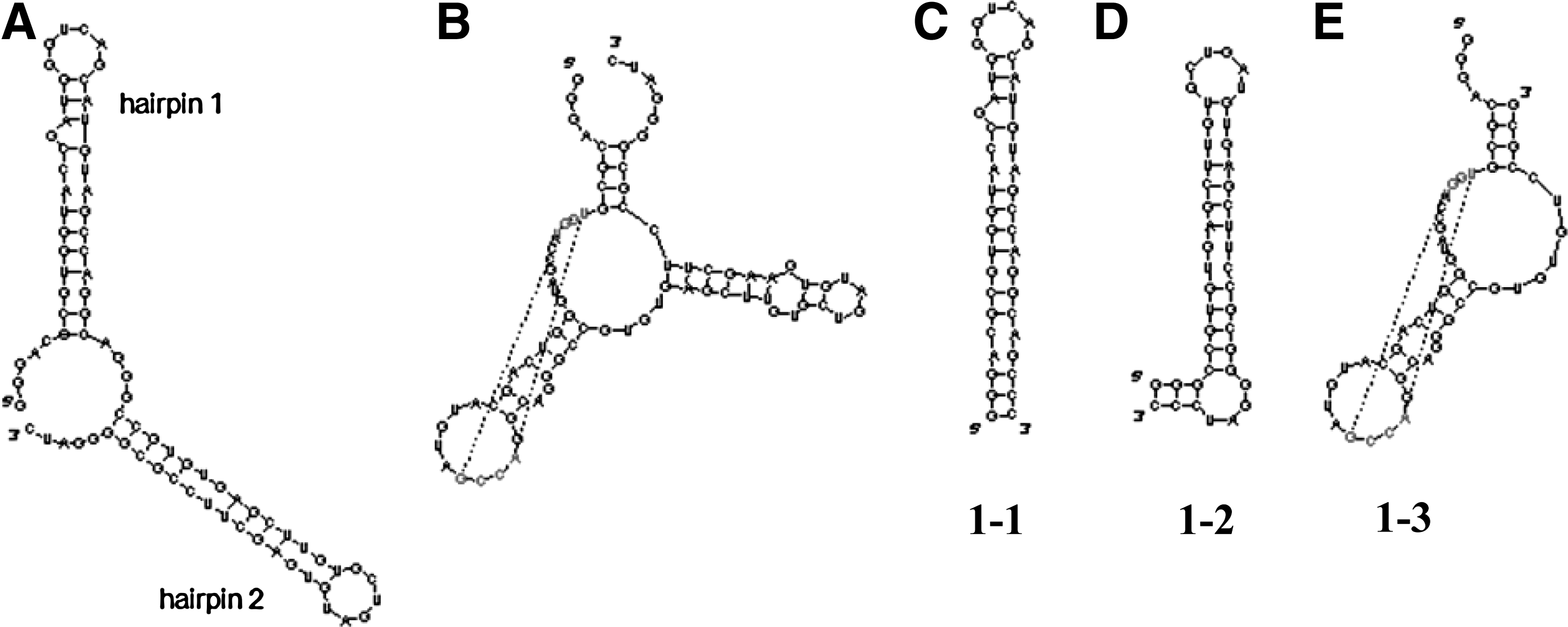
Secondary structure of the clone 1 aptamer and its truncated forms.
5′ Fixed region, GGGACGCGTGGTACC; 3′ fixed region, AGCTTCCGCGGGGATC. Underlined sequences indicate conserved domains. Twenty-nine clones were sequence analyzed after the 10th cycle. Four sequences were finally detected. The most abundant sequence was aptamer clone 2, which shows 24 times out of 24 clones. Multiplicity is given in parentheses.
SPR technique was used to determine the binding affinities. C-terminal biotinylated peptides were loaded on a streptavidin-immobilized CM5 sensor chip using the amine coupling method. Each experiment was triplicated and averaged. Values indicate average and 1 standard deviation of at least 3 experiments.
Selectivity ratio defined as Kd (of other peptide)/Kd (of cognate peptide) is given in parentheses.
SPR, surface plasmon resonance.
SPR technique was used to determine the binding affinities. C-terminal biotinylated peptides were loaded on a streptavidin-immobilized CM5 sensor chip using the amine coupling method. Each experiment was triplicated and averaged. Values indicate average and 1 standard deviation of at least 3 experiments.
Finally, the binding affinities and selectivities of the clone 1 aptamer were compared with those of 2 commercially available antibodies. We attempted to generate polyclonal antibodies against the 14-aa H3R8Me2sym peptide. However, after several intraperitoneal injections of the peptide (100 μg/kg), the titer showed extremely low levels of the antibody as a result of a poor immune response. As the anti-H3R8Me2sym antibody was not available, a direct comparison of its propeties to those of the aptamers against this specific modified peptide was not possible. Instead, binding affinities of 2 available antibodies (anti-H3K9Me2 and anti-H3 nonmodified peptide) were simultaneously determined against the 3 typically modified and nonmodified peptides H3R8Me2sym, H3K9Me2, and nonmodified H3. The results were compared with that of the aptamer. First, no difference exists between the binding affinities of the aptamer and the antibodies. Binding of the antibodies against H3K9Me2 and nonmodified H3 were observed to have Kd values of 7 and 19 nM, respectively. The affinities are similar to that of the aptamer (12 nM). Second, the selectivity of aptamer binding to the target peptides was compared with those of the antibodies. To facilitate the comparison, selectivity ratios (Kd of other modified peptide/Kd of cognate peptide) were calculated (Table 4). Selectivity ratios of the aptamer were determined to be 3.5–8.1 in comparison to 3–4.7 and 2.8–3.7 for anti-H3 nonmodified and anti-H3K9Me2 antibody, respectively. The results show that the aptamer has a larger selectivity ratio than that of the antibodies, despite the fact that symmetric dimethylation on Arg is quite difficult to detect relative to trimethylation on Lys or nonmodified peptides. The findings suggest that the aptamer might be useful as an alternative to antibodies in a tool designed to detect epigenetically modified histones.
Selectivity ratio defined as Kd (of other peptide)/Kd (of cognate peptide) is given in parentheses. The SPR technique was used to determine the binding affinities. Each experiment was triplicated and averaged. Values indicate average and 1 standard deviation of at least 3 experiments.
Conclusion
Epigenetically modified N-terminal histone peptides, especially H3, are important signals for the target genes in chromatin. Therefore, specific tools for detection of these epigenetic modifications in histone H3 N-terminal peptides should have potential value in diagnostic and therapeutic systems. As an alternative to antibodies, RNA aptamers display comparable binding affinities and selectivities against various biologically relevant targets. In recent studies probing the mutual effects of multiple modifications in histone H3 N-terminal peptide, we found that phosphorylation at S10 is totally abolished by symmetric dimethylation at the neighboring R8. Thus, the dimethyl modification could be blocking the signal for phosphorylation and, as a result, play an important role in gene activation. In the effort described earlier, 14-aa peptides that mimic the H3R8Me2sym peptide were prepared in biotinylated forms and subjected to 10 SELEX selection cycles to afford 4 aptamer clones. One aptamer in this group displayed a 12 nM binding affinity and a reasonable selectivity for the cognate peptide. This aptamer is composed of 2 hairpins, both of which might be involved in recognition of the cognate peptide. Finally, the binding affinity and selectivity of the aptamer were compared with those of 2 commercially available antibodies against differently modified histone H3 peptides. The aptamer was found to have a comparable affinity and a much greater selectivity than the antibodies. These findings suggest that aptamers could be potentially selected against any epigenetically methylated peptide, in which the modification is relatively small in comparison to those produced by phosphorylation and acetylation.
Footnotes
Acknowledgments
Financial support of this work was provided by grants from Ministry of Knowledge and Economy (Grant No. 1003211-2010-13) and the Ministry of Science and Education of Korea (Grant No. 20100000297).
Author Disclosure Statement
No competing financial interests exist.