
Abstract
Duchenne muscular dystrophy (DMD) is a lethal X-linked inherited disease caused by mutations in the dystrophin gene and consequent lack of dystrophin in the skeletal, cardiac, and smooth musculature and in the nervous system. Patients die during their mid-twenties because of severe muscle loss and life-threatening respiratory and cardiac complications. The splicing modulation approach mediated by antisense oligonucleotides can restore the production of a partially functional quasi-dystrophin in skeletal muscles. We recently showed that a chronic, 12-month treatment with phosphorodiamidate morpholino oligomers efficiently restored dystrophin in widespread skeletal muscles and led to normal locomotor activity indistinguishable from that of dystrophin-expressing C57 mice. However, no detectable dystrophin expression was observed in the hearts of treated mice. In the present study, histological analyses show a more severe cardiac pathology compared with untreated animals in the face of enhanced locomotor behavior. This observation implies that the increase in locomotor activity of treated mdx mice may have a paradoxical detrimental effect on the dystrophic heart. In the context of skeletal muscle-centric therapies for DMD, our data suggest that particular vigilance should be instigated to monitor emergence of accelerated cardiac dysfunction.
Introduction
Materials and Methods
Administration of PMO to mdx mice
PMO (GeneTools) with the sequence 5′-GGCCAAACCT CGGCTTACCTGAAAT-3′ (Gebski et al., 2003) was used to induce dystrophin exon 23 skipping. PMO dissolved in sterile saline was injected via the tail vein in C57BL/10ScSn-Dmdmdx (mdx) male mice starting at 6 weeks of age. Briefly, 2 dosing regimens were used: 5 cycles of 4 weekly injections of 5 or 50 mg/kg PMO per cycle were administered to mdx mice. Animals were sacrificed at 50 weeks after the first injection (Malerba et al., 2011). Animals were bred in-house. Food and water were provided ad libitum. Mouse husbandry and experimentation adhered to statutory United Kingdom Home Office regulatory, ethical, and licensing procedures and the Animals (Scientific Procedures) Act 1986 (project license: PPL 70/7008).
Histology and immunohistochemistry
Cardiac muscles or liver necropsies were harvested, mounted, and subjected to tissue sectioning. Ten-micrometer-thick transversal sections were placed on coated glass slides (VWR) and stored at −80°C before use. A standard protocol for hematoxylin and eosin staining was used to measure morphometric parameters as the septolateral area and diameter of the left ventricle and the lateral wall thickness of the left and right ventricles. SigmaScan Pro image analysis software (Systat Software) was used for the analysis. Immunofluorescence for dystrophin [P6, rabbit polyclonal, 1:400 (Sherratt et al., 1992)], laminin (a-Merosin, mouse monoclonal, 1:1000; Sigma), and collagen VI (ab6588 rabbit polyclonal, 1:200; Abcam) were performed as previously described (Malerba et al., 2009). For dystrophin and von Willebrand factor costaining, the mouse monoclonal antibody NCL-DYS2 (1:50; Novocastra Laboratories) in conjunction with the rabbit polyclonal anti-human von Willebrand factor antibody (1:1000; Dako, A0082) was used. Goat anti-rabbit Ig (Alexafluor 568; Invitrogen) or rabbit anti-mouse IgG (Alexafluor 488; Invitrogen) was used as secondary antibody. The percentage of the area stained for collagen VI in cryosections was measured in the largest section of the cardiac muscle including the ventricles using SigmaScan Pro image analysis software. Images were captured and analyzed using identical parameters of exposure, saturation, and gamma levels between treated and untreated specimens. Endogenous intracellular IgG staining was evaluated using rabbit anti-mouse IgG (1:100, Alexafluor 488; Invitrogen). Nuclei were counterstained with 4′,6–diamidino–2–phenylindole (DAPI). The intensity of dystrophin staining, performed with the P6 rabbit antibody previously reported, was analyzed by Metamorph software as reported by Arechavala-Gomeza et al. (2009) and Malerba et al. (2011). Serial sections stained for laminin were used to normalize the signal level.
Statistical analyses employed
Data are expressed as mean±standard error. Two-tailed parametric t-test was used for the analyses as indicated. All the statistical analyses were performed with GraphPad Prism software (version 4; GraphPad software Inc.).
Results and Discussion
Starting at 6 weeks of age, mdx mice were intravenously injected via the tail vein on a regular basis with PMO to induce exon 23 skipping over a 50-week period as described by Malerba et al. (2011). Two doses of PMO were tested: 5 mg/kg/injection (low dose [LD]) and 50 mg/kg/injection (high dose [HD]). The injection regimen was 5 cycles and 10 weeks long, each consisting of 4 weekly injections followed by a 6-week rest period of no treatment. Animals were sacrificed for analysis at 6 weeks after the last injection. The rationale underlying the choice of these dosing regimens is that repeated cycles of LD PMO injections may represent an efficient therapeutic approach for DMD, allowing significant dystrophin accumulation during the treatment. Indeed, similar doses have been recently used in a United Kingdom–based clinical trial (NTC00844597). Although mdx mice are not considered an ideal model for dystrophin-deficient cardiac pathology, they do develop heart tissue damage that leads to an extensive dilated cardiac myopathy later in life (Quinlan et al., 2004; Jearawiriyapaisarn et al., 2010). To verify the absence of dystrophin expression in cardiac muscles of animals treated with LD or HD regimens, we used a recently validated, very sensitive, semiquantitative intensity analysis (Arechavala-Gomeza et al., 2009; Kinali et al., 2009). The level of dystrophin expression in the hearts of mdx mice treated with LD and HD of PMOs was similar to the baseline of the protein detected in untreated mdx mice (Fig. 1). A similar outcome was observed after short- (Lu et al., 2005; Alter et al., 2006; Malerba et al., 2009) and long-time (Wu et al., 2011) systemic administration experiments and even when intracardiac injection of PMO was performed (Vitiello et al., 2008). The mechanisms whereby AOs are effectively internalized in skeletal myofibers but not in cardiomyocytes remain to be defined. A possible explanation is that pathological sarcolemmal permeability in dystrophin-deficient skeletal myofibers, which leads to high serum levels of creatine kinase and other muscle enzymes, also facilitates the entry of AOs, and that this effect is absent in cardiac muscles, where cell membrane damage is much less pronounced. Although voluntary exercise of mdx mice can induce structural remodeling in the cardiac muscles (Costas et al., 2010), in the PMO-treated mice examined here, we did not observe a similar outcome. Morphometric parameters useful to identify an advanced state of cardiomyopathy, such as the septolateral area, diameter of the left ventricle, and the lateral wall thickness of the left and right ventricles, were not modified after chronic PMO treatment (Fig. 2). This outcome suggests that the cardiac muscles of PMO-treated mice did not undergo a structural remodeling after increased activity, despite the absence of dystrophin. A recent study utilizing various models of dystrophin or utrophin replacement in mdx mice has suggested that the functional rescue of respiratory muscles may have a beneficial effect on cardiac muscles (Crisp et al., 2011). In the present study, we have previously reported that after 1 year of treatment with LD or HD PMO regimens, the diaphragm of mdx mice expressed about 40% and 60% of dystrophin-positive fibers, respectively. Morphological improvements and a clear delay of the pathology in diaphragm were also observed, particularly after HD treatment (Malerba et al., 2011). Importantly, we also detected dystrophin expression in smooth muscle cells of blood vessels (Fig. 3). The rescue of vascular bed dystrophin expression is also known to ameliorate mdx dystrophic pathology (Ito et al., 2006) and may induce a beneficial effect on the cardiac muscles. Finally, the restoration of a quasi-dystrophin missing the exon 23 rescues the neuronal nitric oxide synthase (nNOS) expression at the sarcolemma (Lu et al., 2003; Malerba et al., 2009). Nitric oxide produced in skeletal muscles is a highly diffusible gaseous free radical that can be transported by the blood to organs and tissues where no dystrophin is expressed (BREDT, 2003). All these factors may have contributed to the protection against a structural remodeling of the heart. However, when we carefully analyzed the histology of cardiac muscles, we observed a substantial increase in fibrosis as judged by collagen type VI deposition in treated mice compared with both the untreated age-matched mdx and C57 mice (Fig. 4A). Both LD and HD PMO-treated mice presented a statistically significant increase in fibrotic areas expressing collagen VI (P<0.001 and P<0.05 for LD and HD, respectively) when compared with untreated mdx mice (Fig. 4B). Further, the examination of intracellular immunoglobulin G accumulation, which is an indicator of pathological disruption of membrane integrity and cardiomyocyte damage, revealed that HD-treated mice exhibited both localized and extended zones of acute cardiac injury that were reduced or absent, respectively, in untreated mdx or in control C57 mice (Fig. 4C). Such zones of intracellular IgG were mainly localized on the walls of the left ventricle. The area covered by intracellular accumulation was significantly larger in HD-treated mice compared with LD-treated or untreated mice (Fig. 4D). A very small amount of dystrophin in the heart (ie, <2%) was found to prevent cardiomyopathy in long-time PMO-treated mdx mice (Wu et al., 2011). A different dosing regimen (fortnightly injection of 60 mg/kg compared with our study of 5 cycles of 4 weekly injections of 50 mg/kg and 6 weeks of rest) and the younger mice chosen for the injections (4–5-week-old compared with the 6–7-week-old mice used in our study) may account for the difference in dystrophin expression and outcomes observed. Further, the higher amount of dystrophin detected in skeletal muscles of mice treated with our dosing regimens significantly increased the activity of treated mice. A similar effect was not reported in the study by Wu et al. (2011). In conclusion, the exacerbated damage we observed in the heart of PMO-treated mdx mice may be the outcome of competing effects. On the one hand, the beneficial partial rescue of respiratory muscles, vascular bed and nNOS expression in skeletal muscles counteracted by the detrimental effect of an increased locomotor activity, increased cardiac workload and local absence of dystrophin on the heart. The naked AO-mediated induction of exon skipping in the dystrophin RNA is a therapy for DMD, which is potentially a lifelong treatment: translating to a licensed medicine, DMD patients would require many years of chronic AO administration in. The lack of dystrophin in the cardiac muscle potentially over such a chronic period leads to concerns that therapies for DMD focused primarily on skeletal muscle, such as the AO-mediated dystrophin restoration, may be associated with increased risk of pathology in cardiac muscle. In AO-treated DMD patients, early diagnosis of deteriorating cardiac function, careful follow-up, and timely treatment (eg, with angiotensin-converting enzyme (ACE) inhibitors, antioxidant beta-blockers, or corticosteroids) may effectively minimize any worsening of dystrophic disease in the heart (Ishikawa et al., 1999; Markham et al., 2008). Looking to the future, a second generation of AO linked to cell-penetrating peptides, PPMOs, may be used in clinical trials if issues of adverse general toxicity and immunoreactivity to peptide sequences can be rendered acceptable (Amantana et al., 2007; Moulton and MOULTON, 2010). These new PPMO compounds have been demonstrated to effectively enter the cardiomyocytes and represent a possible future evolution for the AO-mediated therapy for DMD (Wu et al., 2008; Yin et al., 2008; Jearawiriyapaisarn et al., 2010).

Morphometric quantitation of staining intensity in immunolabeled cryosections of treated mdx cardiac muscles using Metamorph software shows no dystrophin expression. Images of dystrophin-stained heart section reported in the upper panels represent the output of the software. As illustrated by the graph, after 50 weeks of PMO administration, dystrophin expression in LD- and HD-treated cardiac muscles was similar to the baseline of mdx mice. The values of dystrophin intensity were normalized to those of laminin intensity in serial stained sections (mean±SEM; n=4–8; t-test: ns, not significant). PMO, phosphorodiamidate morpholino oligomer; HD, high dose; LD, low dose; SEM, standard error of the mean. Color images available online at www.liebertonline.com/nat
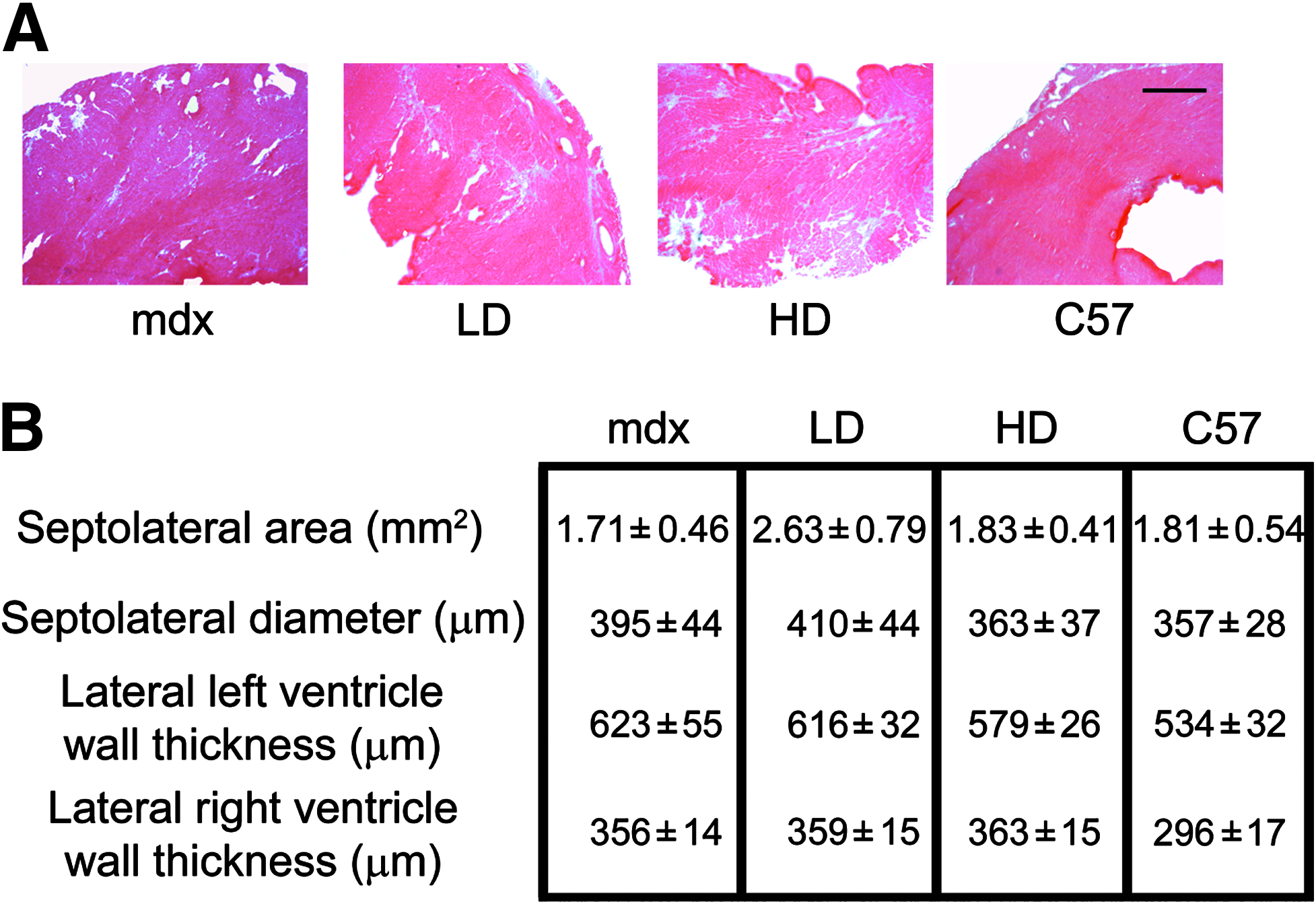
Increased activity due to PMO treatment does not induce structural remodeling of the heart. Morphometric parameters were analyzed in transversal sections of cardiac muscles in treated and untreated mice.
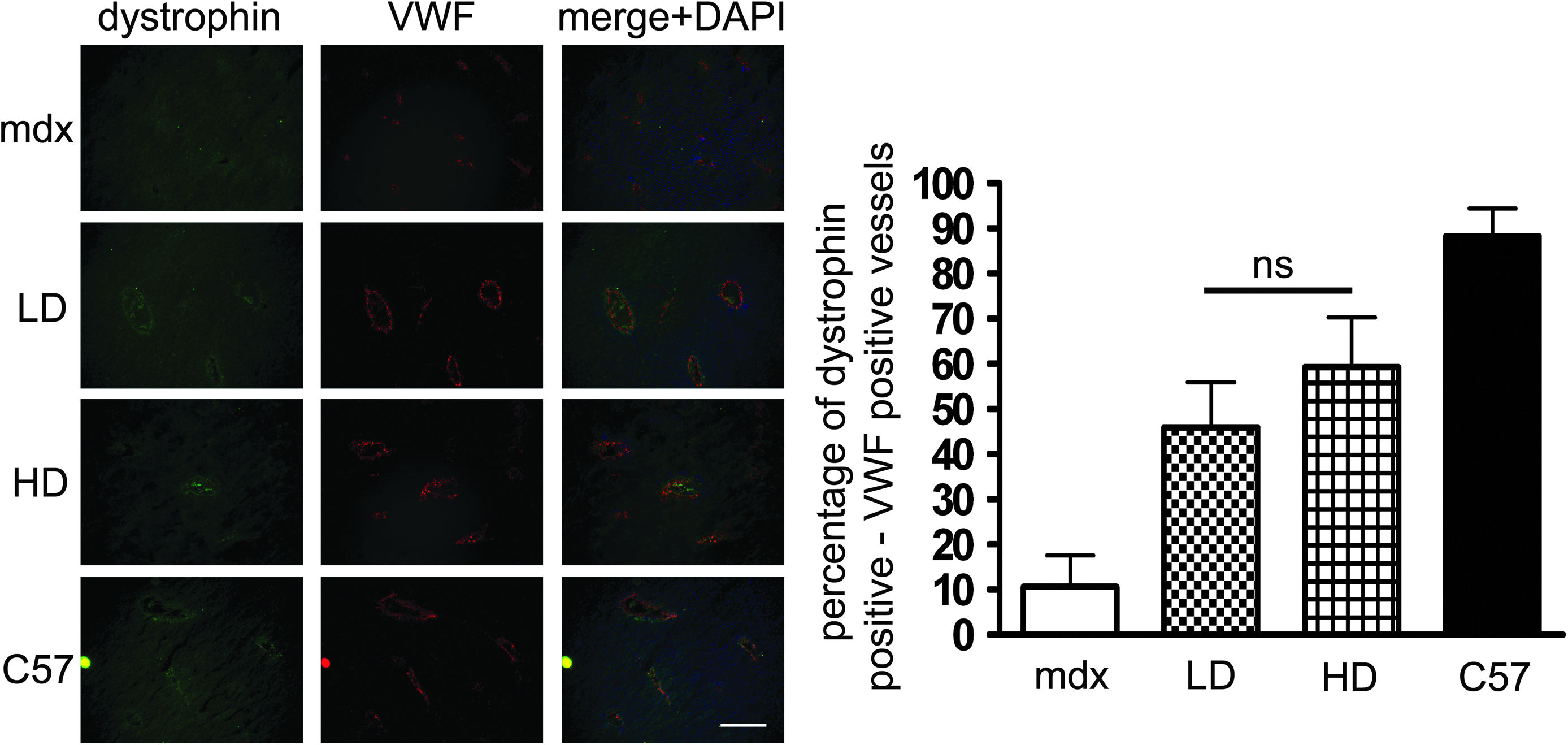
Smooth muscle cells of blood vessels expressed dystrophin after both LD and HD PMO treatment. Serial sections of liver were stained for dystrophin and von Willebrand factor (VMF).

Lack of protection by dystrophin expression in heart may worsen cardiomyopathy in mdx mice.
Footnotes
Acknowledgments
The authors acknowledge the other members of the MDEX Consortium for helpful input and advice. This work was supported by the Muscular Dystrophy Campaign, Muscular Dystrophy Ireland, the EC Clinigene Network of Excellence, the Medical Research Council, and the United Kingdom Department of Health. L. Boldrin was funded by the Muscular Dystrophy Campaign (grant number RA3/776) held by Jennifer Morgan.
Author Disclosure Statement
No competing financial interests exist.