
Abstract
Structural modifications could provide classical small interfering RNA (siRNA) structure with several advantages, including reduced off-target effects and increased silencing activity. Thus, RNA interference (RNAi)-triggering molecules with diverse structural modifications have been investigated by introducing variations on duplex length and overhang structure. However, most of siRNA structural variants are based on the linear duplex structure. In this study, we introduce a branched, non-linear tripartite-interfering RNA (tiRNA) structure that could induce silencing of multiple target genes. Surprisingly, the gene silencing by tiRNA structure does not require Dicer-mediated processing into smaller RNA units, and the 38-nt-long guide strands can trigger specific gene silencing through the RNAi machinery in mammalian cells. tiRNA also shows improved gene silencing potency over the classical siRNA structure when complexed with cationic delivery vehicles due to the enhanced intracellular delivery. These results demonstrate that tiRNA is a novel RNA nanostructure for executing multi-target gene silencing with increased potency, which could be utilized as a structural platform to develop efficient anticancer or antiviral RNAi therapeutics.
Introduction
RNA
Since the first description of RNAi phenomenon in Caenorhabditis elegans with 600∼2,000-bp-long dsRNAs, researchers reported successful gene-specific silencing in various eukaryotic species. However, it was not possible to detect specific gene silencing in mammalian cell lines due to the long dsRNA-mediated non-specific interferon response, until Tuschl and colleagues demonstrated 19-bp-long synthetic siRNA as a potent RNAi trigger in mammalian cells without inducing interferon response (Elbashir et al., 2001a; Elbashir et al., 2001b). Since then, siRNA-mediated RNAi in mammalian cells has been regarded as a promising functional genomics tool and future therapeutic platform owing to their specific gene silencing activities for unlimited target genes.
Initially, RNAi-triggering molecular structure was designed to have a 19-bp duplex region with 2-nt 3′ overhang on both ends, which mimics the Dicer-cleavage product of long dsRNA (Elbashir et al., 2001a; Elbashir et al., 2001c). Follow-up studies, however, have shown that variations on the original siRNA structure are possible (Chang et al., 2011). Importantly, these siRNA structural variants not only expanded the structural diversity repertoire of RNAi triggers, but also came with additional benefits such as reduction of non-specific effects triggered by classical siRNAs, modulation of innate immune stimulation, enhanced cellular delivery, and even improved gene silencing potency (Chang et al., 2011). These observations justify the efforts to explore diverse siRNA structural variants and to test their potential advantages over the classical siRNA structures. However, most of the siRNA structural variants studied have linear structure, and branched, non-linear structures as RNAi triggers have not been explored.
Drug development using RNAi technology has been actively pursued in many fields, including viral infection and cancer. RNAi-mediated inhibition of viral replication has been achieved transiently by siRNA or stably by vector-expressed short hairpin RNA (Wilson and Richardson, 2006). However, in spite of the initial success in the inhibition of viral replication, targeting viruses using single siRNA has limitations, because viruses could escape from the RNAi by rapid introduction of mutations within the viral genome (Das et al., 2004). Single nucleotide substitution or deletion of siRNA target within the viral genome was sufficient to escape from the sequence-specific RNAi. To reduce the chance of viral escape, it is necessary to simultaneously target multiple regions of viral genome by introducing multiple siRNAs (Boden et al., 2003; Das et al., 2004; Westerhout et al., 2005).
This strategy, termed combinatorial RNAi (Grimm and Kay, 2007), is also desired for RNAi-based anticancer therapeutics development. For example, simultaneous targeting of genes involved in cell cycle and apoptosis was more effective in the inhibition of cancer cell proliferation when compared with single gene targeting (Menendez et al., 2004).
In this study, we introduce a branched, non-linear tripartite RNAi-triggering molecular structure, termed tiRNA, which can efficiently inhibit expression of multiple genes simultaneously. Interestingly, tiRNA showed enhanced gene silencing activity compared with conventional siRNAs, due to the increased cellular internalization by cationic delivery vehicles. To our surprise, the long antisense strands of tiRNA triggered effective RNAi without Dicer processing. Therefore, our results provide a simple, synthetic RNA nanostructure for combinatorial RNAi, and add another example in the growing list of novel RNAi-triggering molecular structures, which are structurally and mechanistically distinct from the classical siRNA.
Materials and Methods
siRNA and tiRNA, and control oligonucleotides
Chemically synthesized RNAs and DNAs were purchased from Bioneer and annealed according to the manufacturer's protocol. Sequences of RNAs and DNAs used in experiments are shown below.
siRNA
siDBP sense: 5′-UCGAAGACAUCGCUUCUCA(dTdT)-3′
siDBP antisense: 5′-UGAGAAGCGAUGUCUUCGA(dTdT)-3′
siLamin sense: 5′-CUGGACUUCCAGAAGAACA(dTdT)-3′
siLamin antisense: 5′-UGUUCUUCUGGAAGUCCAG(dTdT)-3′
siTIG3 sense: 5′-CUGUCUCAGGCGUUCUCUA(dTdT)-3′
siTIG3 antisense: 5′-UAGAGAACGCCUGAGACAG(dTdT)-3′
siCVA-CRE sense: 5′-AGAGCAAACACCGUAUUGA(dTdT)-3′
siCVA-CRE antisense: 5′-UCAAUACGGUGUUUGCUCU(dTdT)-3′
siCVA-3D1 sense: 5′-UGGUGAUGAUGUAAUUGCU(dTdT)-3′
siCVA-3D1 antisense: 5′-AGCAAUUACAUCAUCACCA(dTdT)-3′
siCVA-3D2 sense: 5′-CCAUGACUCCAGCUGACAA(dTdT)-3′
siCVA-3D2 antisense: 5′-UUGUCAGCUGGAGUCAUGG(dTdT)-3′
tiRNA
First strand: 5′-UGUUCUUCUGGAAGUCCAGUCGAAGACAUCGCUUCUCA-3′,
Second strand: 5′-UGAGAAGCGAUGUCUUCGACUGUCUCAGGCGUUCUCUA-3′,
Third strand: 5′-UAGAGAACGCCUGAGACAGCUGGACUUCCAGAAGAACA-3′
tiRNA-OMe(2)
First strand: 5′-UGUUCUUCUGGAAGUCCAGUCGAAGACAUCGCUUCUCA-3′,
Second strand: 5′-UGAGAAGCGAUGUCUUCGACUGUCUCAGGCGUUCUCUA-3′,
Third strand: 5′-UAGAGAACGCCUGAGACAGCUGGACUUCCAGAAGAACA-3′
tiRNA-OMe(4)
First strand: 5′-UGUUCUUCUGGAAGUCCAGUCGAAGACAUCGCUUCUCA-3′,
Second strand: 5′-UGAGAAGCGAUGUCUUCGACUGUCUCAGGCGUUCUCUA-3′,
Third strand: 5′-UAGAGAACGCCUGAGACAGCUGGACUUCCAGAAGAACA-3′
tiRNA-OMe(6)
First strand: 5′-UGUUCUUCUGGAAGUCCAGUCGAAGACAUCGCUUCUCA-3′,
Second strand: 5′-UGAGAAGCGAUGUCUUCGACUGUCUCAGGCGUUCUCUA-3′,
Third strand: 5′-UAGAGAACGCCUGAGACAGCUGGACUUCCAGAAGAACA-3′
tiRNA-mut
First strand: 5′-UCAAGAACUGGAAGUCCAGUCGAAGACAUCGCUUCUCA-3′,
Second strand: 5′-UGAGAAGCGAUGUCUUCGACUGUCUCAGGCGUUCUCUA-3′,
Third strand: 5′-UAGAGAACGCCUGAGACAGCUGGACUUCCAGUUCUUGA-3′
tiRNA-CVA
First strand: 5′-UCAAUACGGUGUUUGCUCUUGGUGAUGAUGUAAUUGCU-3′,
Second strand: 5′-AGCAAUUACAUCAUCACCACCAUGACTCCAGCUGACAA-3′,
Third strand: 5′-UUGUCAGCUGGAGUCAUGGAGAGCAAACACCGUAUUGA-3′
tiDNA
First strand: 5′-TGTTCTTCTGGAAGTCCAGTCGAAGACATCGCTTCTCA-3′,
Second strand: 5′-TGAGAAGCGATGTCTTCGACTGTCTCAGGCGTTCTCTA-3′,
Third strand: 5′-TAGAGAACGCCTGAGACAGCTGGACTTCCAGAAGAACA-3′
Cell culture and siRNA transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum. Cells were plated in 12-well plates, 24 hours before transfection at 60% confluency in complete medium without antibiotics. siRNAs and tiRNAs were transfected using PEI (Polyplus) (N/P=5) or Lipofectamine 2000 (Invitrogen) (2 μL/1 μg siRNA) reagents following the manufacturer's protocol.
Quantitative RT-PCR
Total RNAs were extracted from cell lysates using the Isol-RNA Lysis Reagent kit (5 Prime). They were then used as templates for cDNA synthesis, which was performed with the ImProm-II™ Reverse Transcription System (Promega) according to the manufacturer's protocol. Target gene expression levels were analyzed by qRT-PCR using a step 1 real-time PCR system (Applied Biosystems) according to manufacturer's protocol. The primer sequences for each gene were as follows:
GAPDH-forward: 5′-GAGTCA ACGGATTTGGTCGT-3′′ GAPDH-reverse: 5′-GACAAGCTTCCCGTTCTCAG-3′ Lamin A/C-forward: 5′-CCGAGTCTGAAGAGGTGGTC-3′ Lamin A/C-reverse: 5′-AGGTCACCCTCCTTCTTGGT-3′ DBP-forward: 5′-CCTCGAAGACATCGCTTCTC-3′ DBP-reverse: 5′-GCACCGATATCTGGTTCTCC-3′ TIG3-forward: 5′-AGATTTTCCGCCTTGGCTAT-3′ TIG3-reverse: 5′-TTTCACCTCTGCACTGTTGC-3′
Native gel electrophoresis
Each siRNA variant was separated on a 10% (w/v) non-denaturing polyacrylamide gel, stained with ethidium bromide (EtBr) and visualized by UV transillumination.
Intracellular uptake analysis
To visualize cellular uptake of siRNA and tiRNA
Plasmids
DNA oligonucleotides corresponding to the CVA24 target mRNA sequences (see below for the DNA sequences) were subcloned into the SpeІ and HindІІІ sites of the pMIR Report-luciferase vector (Ambion).
Luciferase reporter assay
Twenty-four hours after transfection, cells were lysed with the passive lysis buffer of the Dual-luciferase Reporter Assay System (Promega). Luciferase activities were measured using a Victor3 plate reader (PerkinElmer) for both firefly and Renilla luciferase.
Serum stability of siRNA and tiRNA variants
RNA (0.1 nmole)was incubated in 50 μL of 10% fetal bovine serum solution. Seven microliters of each sample was taken at the indicated time points and immediately frozen at −70°C. A 3-μL aliquot of each sample was then separated in a 10% (wt/vol) non-denaturing polyacrylamide gel, stained with EtBr, and visualized by UV transillumination.
Ago-2 incorporation assay
HEK293 cells were transfected with a plasmid encoding FLAG-Ago2 (3 μg per 100 mm dish) using Lipofectamine 2000. Forty-eight hours after transfection, cells were lysed in lysis buffer [50 mM Tris-Cl (pH8.0), 150 mM NaCl] for 30 minutes at 4°C, followed by centrifugation (3200 rpm, 20 minutes, 4°C) to obtain the supernatant. Finally, 500 μL of this cell lysate was incubated with 0.5 pmole of 32P-labeled siRNA mixture or tiRNA for 2 hours at 4°C with gentle disturbance. To this sample mixture, 20 μL of prewashed anti-Flag conjugated agarose bead (Sigma Aldrich) was added and the sample was incubated for 1 hour. The immunoprecipitates were washed, resuspended in lysis buffer, and RNA was extracted by phenol chloroform extraction. For input, after addition of agarose bead the samples were subjected to phenol chloroform extraction. The input and Ago-2 immuno-precipitated RNA were resolved on 10% non-denaturing PAGE gel, transferred to Whatman 3M paper, with the help of gel dryer, and finally exposed to X-ray film.
5′-RACE assay
Eighteen hours after siRNA transfection into HeLa cells, total RNAs were extracted using Isol-RNA Lysis Reagent kit (5 Prime). Three micrograms of total RNAs from each siRNA transfected cells were ligated to 0.25 μg of GeneRacer RNA oligo. The GeneRacer RNA oligo-ligated total RNA was reverse transcribed using GeneRacer oligo dT and SuperScript™ III RT kit (Invitrogen). RNA oligo-ligated mRNAs were amplified using gene-specific primers. The resulting PCR products were cloned into T&A vector (RBC) and sequenced using M13 forward primer.
Lamin gene specific 3′ primer: 5′-CCAGTGAGTCCTCCAGGTCTCGAAG-3′ Lamin gene specific 3′ nested primer: 5′-CCTGGCATTGTCCAGCTTGGCAGA-3′ DBP gene specific primer: 5′-CGGGACAGCACGGCGCGGTAGT-3′ DBP gene specific 3′ nested primer: 5′- CTCCTGGCGCACGGCCACAACTT-3′ TIG3 gene specific 3′ primer: 5′-GGGGCAGATGGCTGTTTATTGATCC-3′ TIG3 gene specific 3′ nested primer: 5′-ACTTTTGCCAGCGAGAGAGGGAAAC-3′
In vitro Dicer cleavage assay
Thirty picomoles of each siRNA or tiRNA(OMe) was cleaved using Turbo Dicer siRNA Generation Kit (Genlantis) for 12 hours (reaction mixtures were made according to manufacturer's protocol). A 2-μL aliquot of each reaction was separated on a 10% non-denaturing PAGE gel, stained with EtBr, and visualized by UV transillumination.
Results
Design of tiRNA structure with multiple gene-silencing functions
We constructed a tiRNA duplex harboring three 19-bp siRNA units directed against Lamin A/C (siLamin), DBP (siDBP), and TIG3 (siTIG3) mRNA, respectively (Fig. 1A). This tiRNA structure was built by annealing three 38-nt single-stranded (ss) RNAs, such that each duplex region contained 19 bp of each siRNA duplex. The structure of tiRNA was designed so that the 5′ end of the antisense strand of each siRNA unit was oriented toward the outside since this arrangement retains the gene silencing activity of the individual siRNA units in longer RNA duplexes (Chang et al., 2007; Chang et al., 2009a). Native gel electrophoresis confirmed that the tiRNA was represented by a single band, which migrated slightly slower than a 38-bp dsRNA (Fig. 1B).
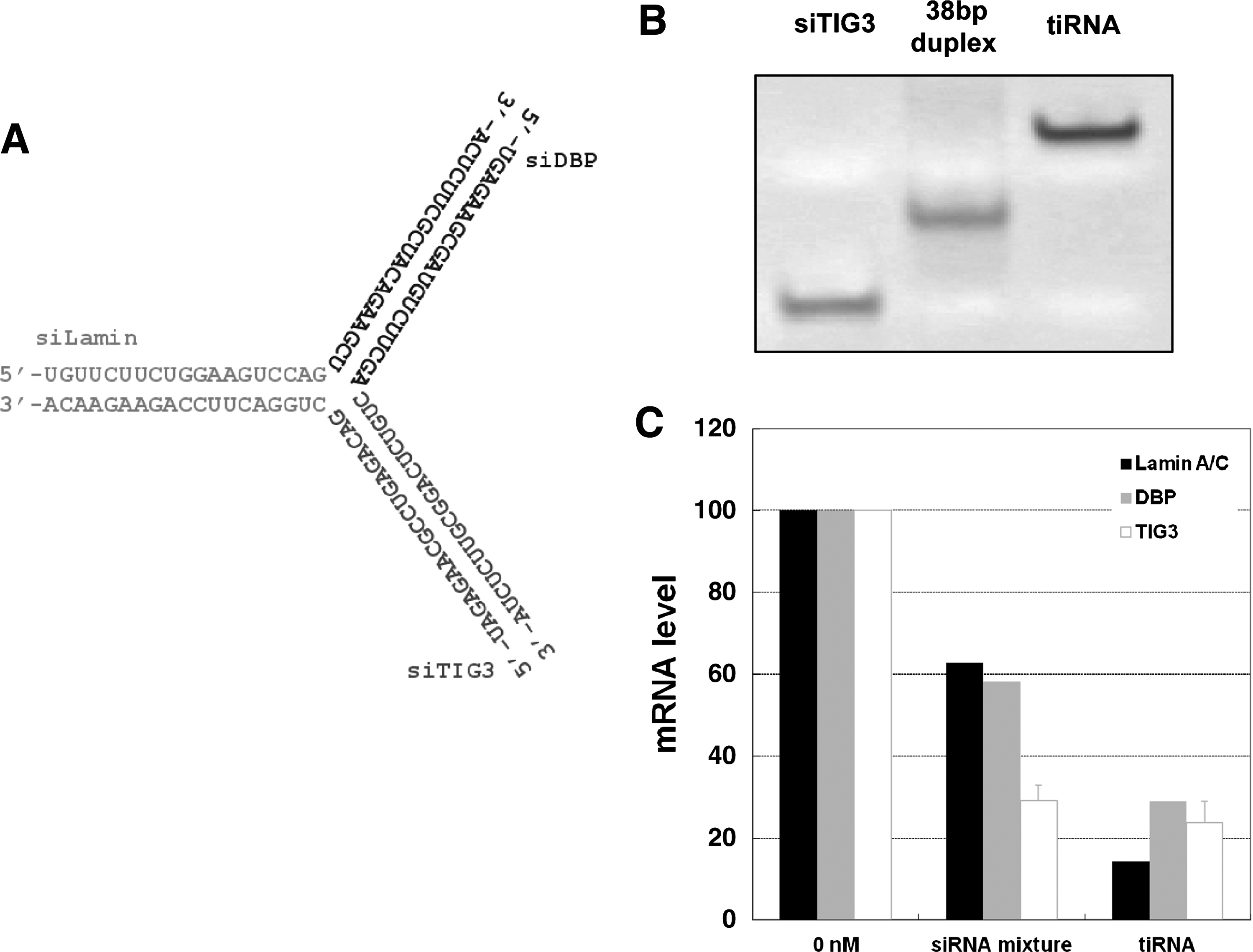
tiRNA structure and activity.
We transfected this tiRNA into HeLa cells using polyethyleneimine (PEI), a widely used transfection reagent for in vitro and in vivo delivery of nucleic acids, including siRNAs (Urban-Klein et al., 2005; AIGNER, 2006). Twenty-four hours after transfection, we measured the gene silencing activity of tiRNA using quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) and compared it to a siLamin/siDBP/siTIG3 mixture. The siRNA mixture induced only marginal gene silencing activity (Fig. 1C), although each siRNA used was a potent one with subnanomolar IC50 values when transfected using Lipofectamine2000 or other conventional transfection reagents (Chang et al., 2009b). This compromised gene silencing by siRNA-PEI complex is likely due to weak complex formation of siRNAs with PEI, which is caused by the small number of negative charges on the 19-bp siRNA (Lee et al., 2010; Mok et al., 2010; Bolcato-Bellemin et al., 2007). In contrast, the tiRNA-PEI complex induced more potent gene silencing activity than the siRNA mixture for all 3 target genes (Fig. 1C). We also tested the gene silencing activity of the tiRNA using Lipofectamine2000, a widely used cationic lipid transfection reagent. Similar to the results obtained using PEI, the tiRNA showed better gene silencing activity than the siRNA mixture for all 3 target genes (Supplementary Fig. S1; Supplementary Data are available online at www.liebertonline.com/nat).
To test whether the gene silencing activity of tiRNA is sequence specific, we synthesized another tiRNA structure with mutation in the seed sequence of siLamin (tiRNA-mut). tiRNA-mut was transfected into HeLa cells and target mRNA levels were analyzed by qRT-PCR. tiRNA-mut completely lost the silencing activity against Lamin mRNA while it maintained silencing activities for the remaining 2 target genes (Supplementary Fig. S2). This result demonstrates that the gene silencing activity of tiRNA is sequence-specific and seed-sequence dependent, like conventional siRNAs.
For RNAi-based inhibition of viral replication, simultaneous targeting of multiple sites within the viral genome is desired to avert the emergence of viral escape due to the high mutation rate (Westerhout et al., 2005; Grimm and Kay, 2007). To test whether tiRNA structures could target 3 different regions of viral genome, we synthesized three 21-bp siRNA (siCRE, si3D1, si3D2) and tiRNA (tiRNA-CVA) structures targeting 3 different regions of the genome of coxsackievirus A24 (CVA24), which is responsible for acute hemorrhagic conjunctivitis, a highly contagious eye disease for which no prevention or treatment is currently available (Jun et al., 2008) (Supplementary Fig. S3A). We also designed three reporter constructs that encode luciferase mRNA bearing target sequences of siCVA-CRE, siCVA-3D1, and siCVA-3D2 at the 3′ untranslated region, respectively. These constructs were then transfected into HeLa cells using PEI along with either siRNA mixture or tiRNA-CVA, respectively, and luciferase activity was measured. Similar to the results in Fig. 1, tiRNA-CVA also show increased silencing activity compared with 21-bp siRNA mixtures for all 3 target sequences (Supplementary Fig. S3B). Taken together, these results demonstrate that the tiRNA structure not only executes triple target gene silencing, but also enhances silencing efficiency over conventional 21-bp siRNAs when complexed with cationic polymer- or lipid-based transfection reagents.
tiRNA triggers gene silencing through the RNAi pathway utilized by conventional siRNAs
Next, to test whether tiRNA triggers gene silencing through the RNAi pathway utilized by conventional siRNAs, we performed a 5′-RACE (Rapid Amplification of cDNA Ends) assay to analyze the cleavage sites of each target mRNA. 5′-RACE PCR products from tiRNA-transfected cells were similar in size to those from siRNA mixture-transfected cells (Fig. 2a-c). For more detailed analysis of cleavage sites, 5′-RACE PCR products from each reaction were cloned and three independent clones from each reaction were sequenced. Both the 19 bp siRNA mixture and the tiRNA cleaved the target mRNAs at the same location: sites corresponding to 10 or 11 nt downstream of the 5′- end of the antisense strand of each siRNA unit (Fig. 2a-c). These results suggest that the tiRNA triggers target gene silencing through the RNAi pathway utilized by the conventional 19 bp siRNAs.
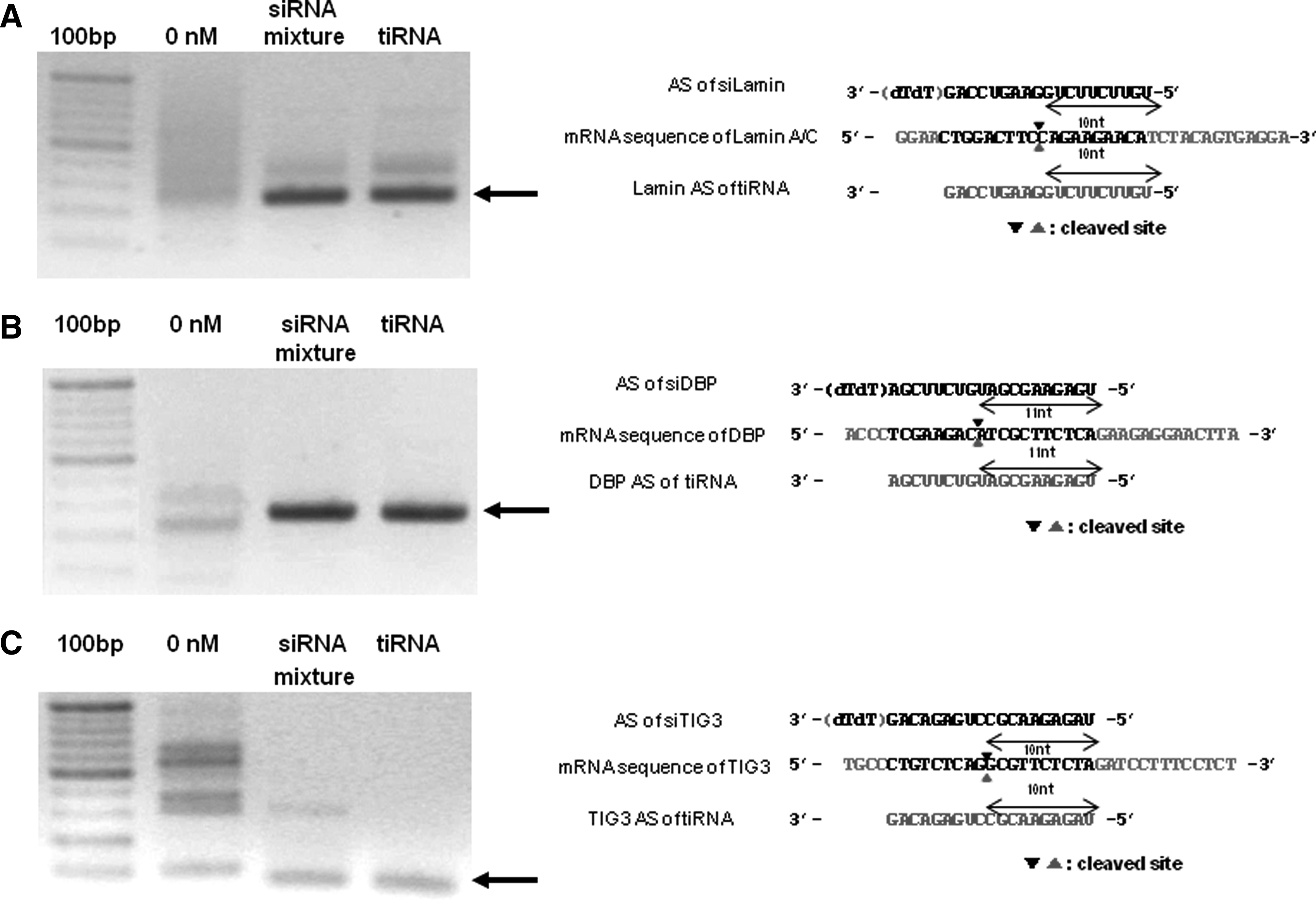
tiRNA works via RNAi mechanism like classical siRNAs. Cleavage sites of target mRNAs were analyzed by sequencing.
tiRNA structure enters the cells with better efficiency than 21bp siRNA
For the development of RNAi therapeutics, efficient delivery of interfering RNA structure into the cell is necessary. However, compared with long nucleic acids such as plasmid DNA, siRNAs bind inefficiently to cationic delivery vehicles such as PEI due to the smaller number of negative charges (Bolcato-Bellemin et al., 2007). Therefore, when the complexes of siRNAs and cationic delivery reagents pass through the cell membrane, the weak interaction between siRNAs and delivery reagents results in the exchange of siRNAs and the negatively charged proteoglycans at the cell surface, resulting in compromised gene silencing (Bolcato-Bellemin et al., 2007). Because tiRNA structure shows better target gene silencing than siRNAs, we wondered if this increased gene silencing efficiency resulted from the enhanced intracellular delivery of tiRNAs owing to the increase in the number of negative charges in the tiRNA structure.
To compare the delivery efficiency of tiRNA and siRNA, we labeled the 3′ end of the sense strand of siTIG3 and tiRNA with fluorescein isothiocyanate (FITC). The FITC-labeled siRNA mixture (siLamin, siDBP, and FITC-siTIG3) and the corresponding FITC-labeled tiRNA were transfected into HeLa cells using PEI and fluorescence intensities were observed by fluorescence microscopy at various time points after transfection. HeLa cells incubated with FITC-labeled siRNA mixture show almost no fluorescence at all incubation times we tested. In contrast, HeLa cells incubated with FITC-labeled tiRNA show scattered green spots in intracellular region, and their intensities were gradually increased with increased incubation times (Fig. 3). These results demonstrate that tiRNA structure is more efficiently delivered into cells for the same incubation times. We also measured activities of siRNA mixture and tiRNA with different transfection times. For all transfection times, more potent silencing activities were observed for tiRNA than for the siRNA mixture (Supplementary Fig. S4). These results demonstrate that tiRNA structures are delivered into the target cells more efficiently than the conventional siRNA, and correlation exists between increased silencing activity and increased delivery efficiency.
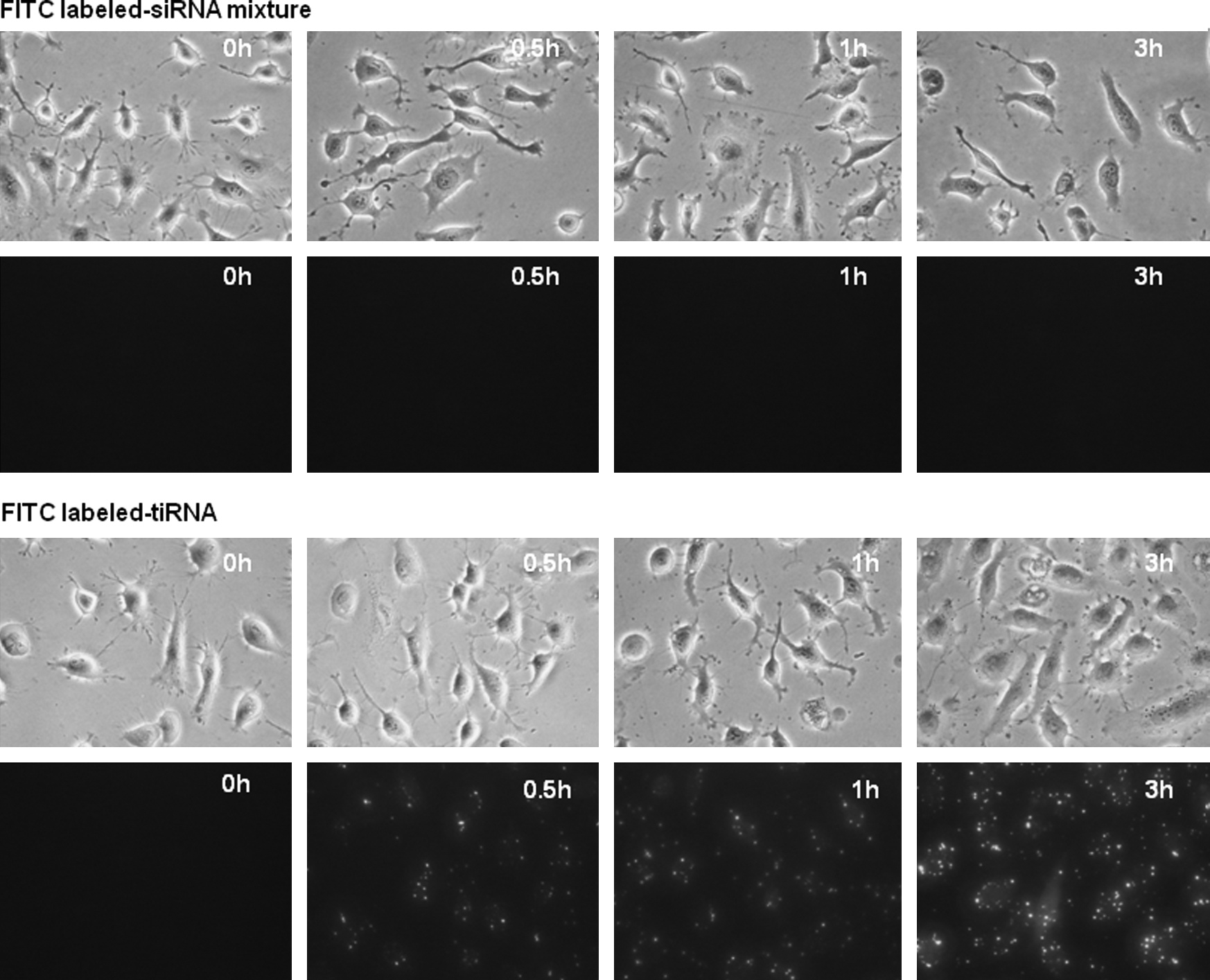
tiRNA is more efficiently delivered into cells than the 19-bp siRNA mixture. The 3′ end of the TIG3 sense strand in a 19-bp siRNA mixture (100 nM each) and a tiRNA (100 nM) were labeled with FITC and transfected into HeLa cell using polyethyleneimine (PEI). The HeLa cells transfected with the 21-bp siRNA mixture showed almost no fluorescence at all time points, but the cells transfected with FITC-labeled tiRNA showed strong fluorescence with scattered fluorescent spots.
The junction structure of tiRNA is nuclease susceptible
Because tiRNA has a unique branched structure, its nuclease susceptibility might be different from that of linear siRNAs. To test the serum nuclease stability of tiRNA structure, tiRNA or siRNA was incubated in 10% serum solution and then visualized by polyacrylamide gel electrophoresis. siRNA was gradually degraded as incubation time increased and was almost undetectable after 24-hour incubation (Fig. 4A). Interestingly, incubation of tiRNA in serum rapidly yielded small dsRNA product with size similar to siRNA, which then showed a similar degradation pattern to that of siRNA (Fig. 4A).
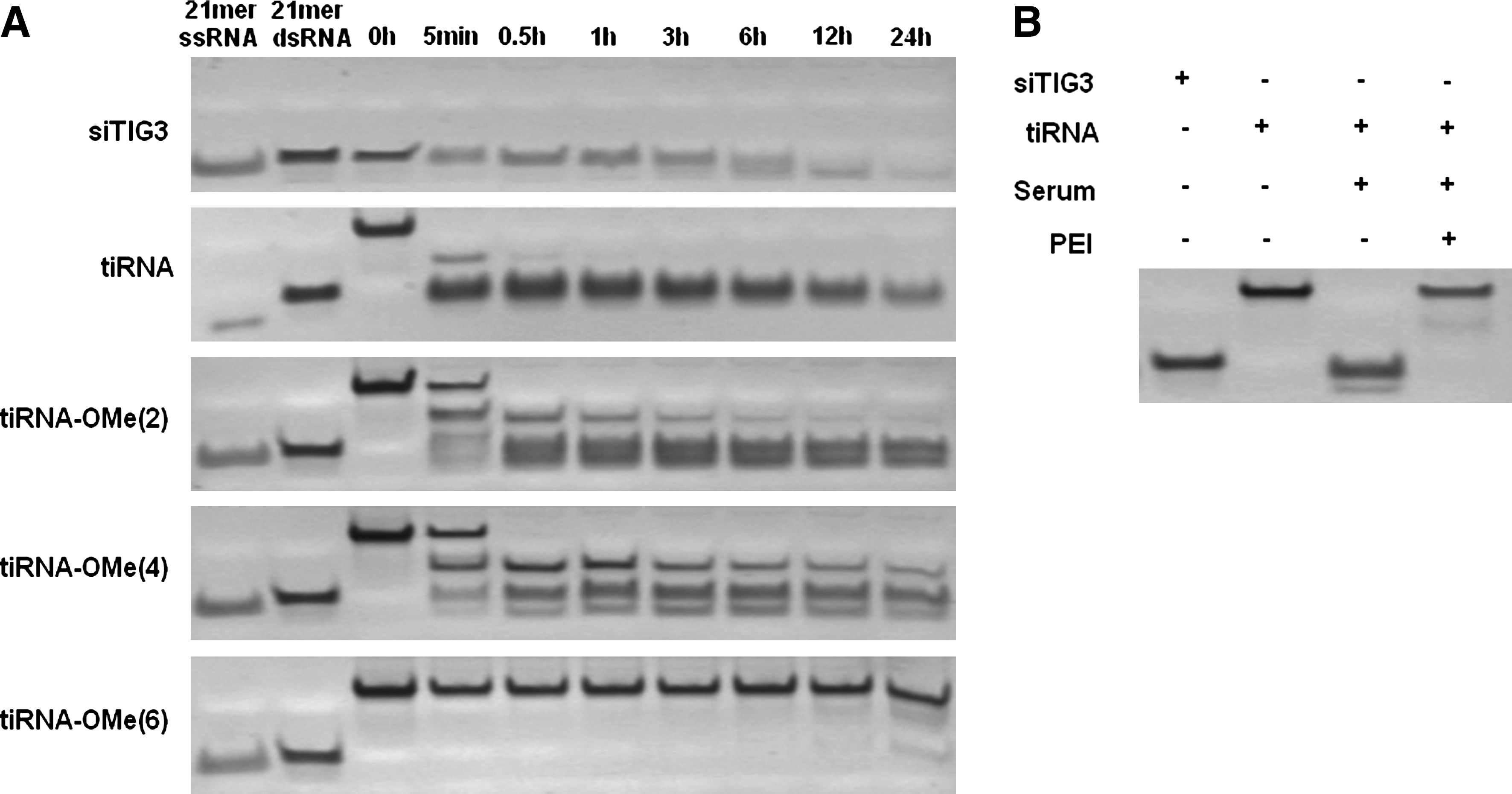
Serum nuclease stability of tiRNA and 2′-OMe modified tiRNAs.
Single-stranded RNA (ssRNA) tends to be more susceptible to nuclease attack compared with dsRNA. Because of the structural constraint in the tiRNA junction structure, base paring near the junction region might be disrupted, and a single-stranded region could be formed. To describe more precisely the junction structure of tiRNA and measure the approximate size of the single-stranded region at the tiRNA junction, tiRNAs modified with nuclease-resistant 2′-OMe modification at their junction region were synthesized. Modification of 2 nt or 4 nt at the junction region with 2′-OMe was not sufficient to prevent the formation of ∼19-bp degradation product (Fig. 4A). However, tiRNA with modification of 6 nt with 2′-OMe at the junction was almost completely resistant to the serum nuclease attack and allowed tiRNA to maintain its intact structure in 10% serum (Fig. 4A). From these results, we hypothesize that tiRNA has a bulge structure at its junction where ∼6-nt single-stranded regions are exposed without base pairing (Supplementary Fig. S5).
It is well known that cationic polymers protect nucleic acids from the attack of nucleases in the serum (Harada et al., 2001). Although tiRNA has a nuclease-susceptible structure, when tiRNA is complexed with PEI, PEI could protect tiRNA junction part from the serum nuclease attack before entering the cells. To test the protection efficiency of PEI, naked tiRNA or tiRNA-PEI complex was incubated with serum, and cleavage patterns were compared. While naked tiRNA was completely cleaved into ∼19bp dsRNAs, tiRNA complexed with PEI was not cleaved into shorter dsRNAs in the serum (Fig. 4B). These results show that tiRNA complexed with PEI is protected from the attack of nucleases in the serum during transfection so that tiRNA enter into the cells intact.
Gene silencing by tiRNA does not require Dicer processing and is executed by long antisense strands
When introduced into cells, tiRNA might be processed by Dicer to yield short dsRNAs (Bernstein et al., 2001). However, while Dicer efficiently processes linear dsRNAs longer than 21∼23 bp, tiRNA has only 19-bp-long dsRNA segments and a central bulge structure; therefore, tiRNA structure might not be a good substrate for Dicer processing. To confirm this hypothesis, tiRNA or siRNA mixture was transfected into HeLa cells, and 24 hours post-transfection, RNAs introduced in the cells were visualized by Northern blot. Consistent with our hypothesis that tiRNA structure might be resistant to Dicer processing, most of the transfected tiRNAs maintained the intact form of 38-nt-long antisense strands (Fig. 5A–C), while they show potent gene silencing activity at the same time point. This result suggests that, unlike long, linear dsRNAs or 27-bp Dicer substrate siRNA, the branched tiRNA structure is not processed by Dicer and maintains 38-nt-long antisense strands in the cells. At the same time, tiRNA shows potent RNAi activity, suggesting that long antisense strand of tiRNA could trigger RNAi without Dicer processing.
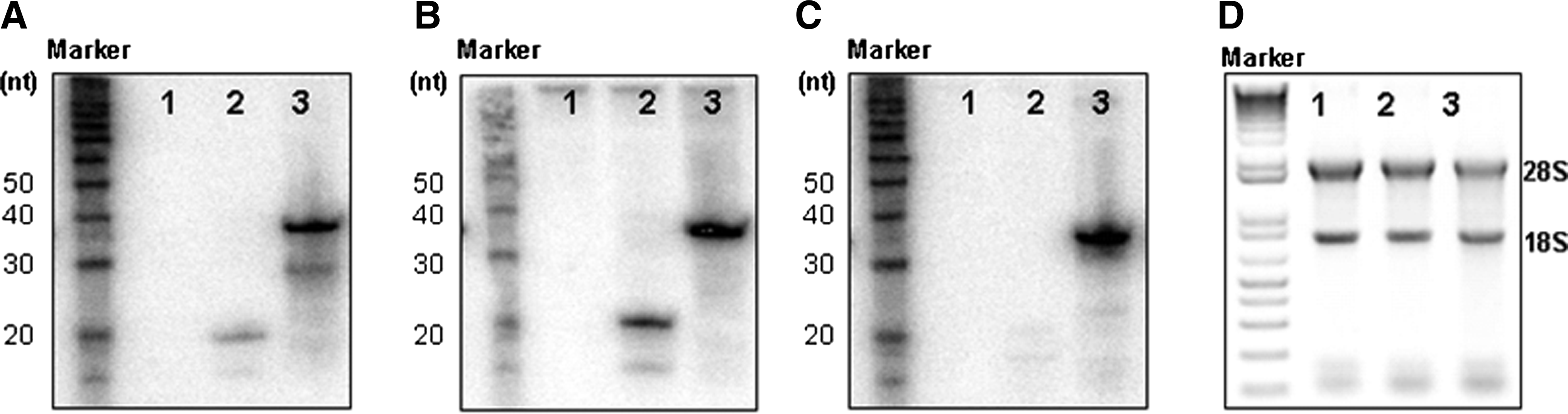
tiRNA is not cleaved into 19-bp dsRNAs and exist as intact form in the cells.
To argue against the possibility that a small amount of Dicer-processed tiRNA, which is not readily visible in Northern blot, might trigger RNAi with increased potency as previously proposed (Kim et al., 2005), we tested the gene silencing activity of tiRNA-OMe(6), the junction-modified, nuclease-resistant tiRNA structure (Fig. 6A). In vitro Dicer cleavage experiment showed that while 27-bp dsRNA are processed into ∼21-bp dsRNAs, tiRNA-OMe(6) was completely resistant to cleavage by Dicer (Fig. 6B).
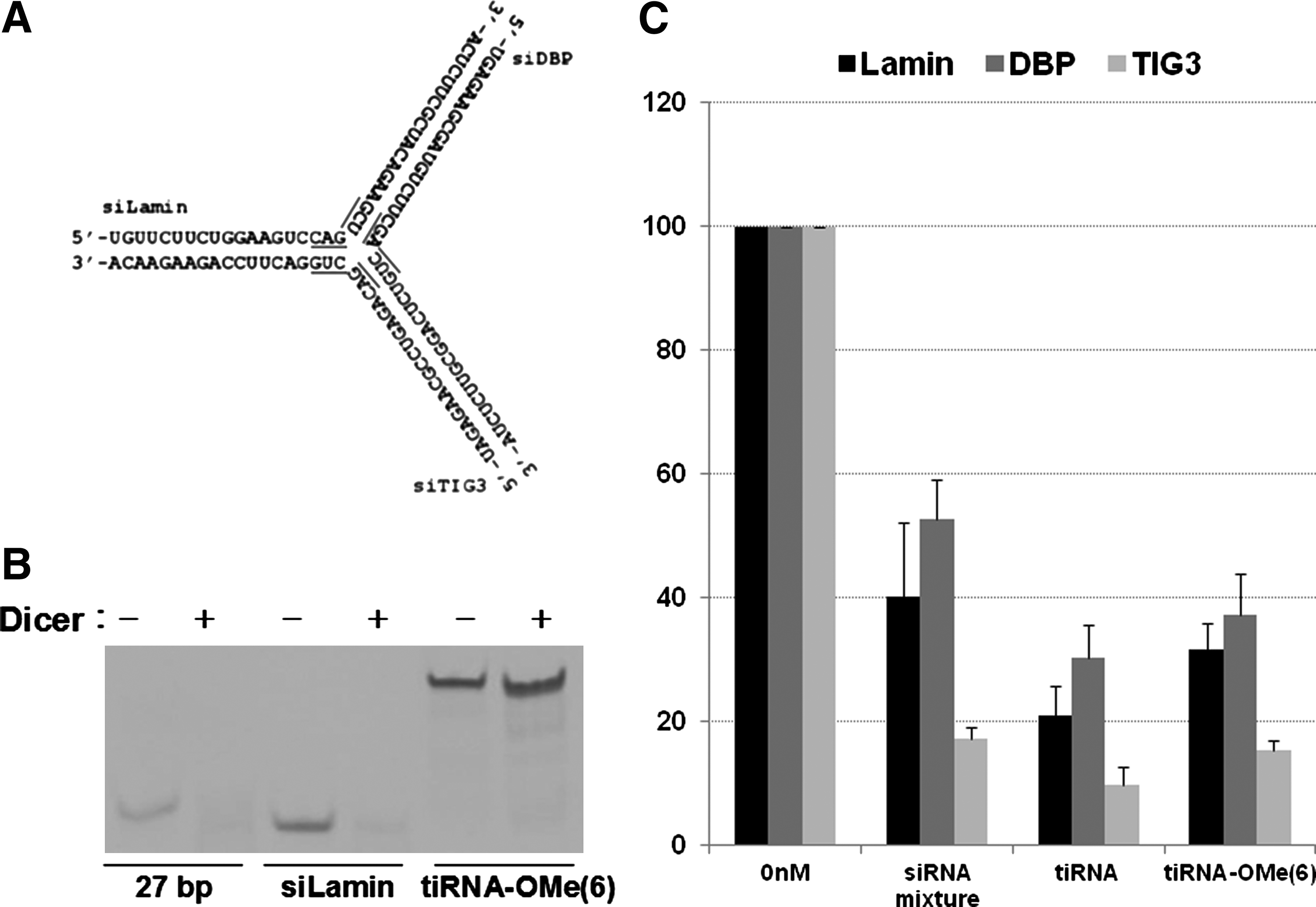
Structure, processing, and activities of tiRNA modified with 2′-OMe RNA.
We then transfected tiRNA-OMe(6) into HeLa cells and measured its gene silencing activity using qRT-PCR. Similar to the unmodified tiRNA structures, tiRNA-OMe(6) showed more potent gene silencing activity than the mixture of 19-bp siRNAs (Fig. 6C). Combined together, these results demonstrate that the tiRNA structure can execute efficient gene silencing with their intact 38-nt antisense strand without further processing into small dsRNAs by Dicer.
38-nucleotide-long antisense strand of tiRNA is incorporated into RISC
Although 38-nt-long antisense strands of tiRNA remain intact in the cells without processing by Dicer while maintaining efficient silencing activity, it is not clear whether 38-nt-long antisense strands of tiRNA incorporate into RISC and trigger RNAi. To test whether the long antisense strands of tiRNA are incorporated into RISC, we performed Ago2 incorporation assay with tiRNA. siRNAs, tiRNA, or tiRNA-OMe(6) labeled with 32P was incubated with the lysate of HEK293 cells overexpressing FLAG-tagged Human Ago2 protein. Then, Ago2 was pulled down with anti-FLAG antibody, and RNAs bound to Ago2 protein were analyzed. We observed that the intact 38-nt-long antisense strand of tiRNA and tiRNA-OMe(6) could directly bind to Ago2 protein without further processing into smaller RNAs (Fig. 7). We also confirmed that this incorporation is not a non-specific binding between tiRNA and Ago2 protein using tripartite DNA structure (tiDNA), which has the same structure with tiRNA but made of DNA instead of RNA. Under the same condition, tiDNA did not show any binding to Ago2 (Supplementary Fig. S6). Combined with northern blot analysis and Dicer cleavage assay results (Figs. 5, 6), these results imply that a 38-nt-long antisense strand of tiRNA can be incorporated into an active RISC complex and guide RNAi in mammalian cells without Dicer processing.

38-nt-long tiRNA antisense strands efficiently associate with Ago2. The 32P-labeled siRNA mixture, tiRNA, or tiRNA-OMe(6) was incubated with cell lysate expressing Ago2 protein, and Ago2-bound RNAs were analyzed.
Discussion
Several recent reports on structural modification of siRNA have highlighted the possibilities of novel RNAi triggers with improved features over classical siRNA structure (Chang et al., 2011). In this study, we constructed a branched, non-linear tiRNA, which could simultaneously inhibit expression of 3 target genes with greater potency than the classical siRNA mixture.
Guo and colleagues previously reported the design of Phi29 RNA-based multi-target RNA structure (Khaled et al., 2005). While both structures execute multi-target gene silencing, the structural design of tiRNA is conceptually different from the Phi29 RNA-based multifunctional RNA. We designed the tiRNA structure using only siRNA unit sequences and did not include any linker sequence. In contrast, Phi29 RNA-based multifunctional RNA structure harbors long extra linker sequences unrelated with their target genes to connect siRNA units. Because it is desired for therapeutic RNA molecules to be as short as possible to reduce production cost and maximize the quality of synthesis, we believe tiRNA is therapeutically more relevant structural platform than Phi29-based RNA structure.
The most intriguing finding was that tiRNA is not processed by Dicer into short dsRNA and guide RNAi with the 38-nt-long antisense strands. The 38-nt-long antisense strands from tiRNA was efficiently incorporated into RISC and cleaved target mRNA at 9–10 nt from its 5′ end of antisense strand, which is a unique feature of the RNAi mechanism. A recent study also reported RNAi-triggering molecular structure that is not processed by Dicer (Salomon et al., 2010). While these findings seem contradictory to the currently accepted RNAi mechanism, the execution of RNAi by long antisense strands is consistent with a recent structural study of Ago2/antisense strand/target mRNA ternary complex, which showed that the 3′ end of the antisense strand was released from the PAZ domain (Wang et al., 2009; Sashital and Doudna, 2010). While further studies are required to understand how RNAi is triggered by long antisense strand RNA, these results emphasize the structural flexibility of the mammalian RNAi machinery.
In conclusion, tiRNA structure expands the structural diversity repertoire of siRNAs suitable for multi-target gene silencing, with improved features over classical siRNAs and existing multi-target RNA structures. We believe that the tiRNA structure has great potential to become a useful structural platform in RNAi-based multi-target gene silencing to develop efficient antiviral or anticancer therapeutics development.
Footnotes
Acknowledgments
This work was supported by a Global Research Laboratory grant from the Ministry of Education, Science, and Technology (MEST) of Korea (no. 2008-00582).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.