
Abstract
The serine protease inhibitor (serpin) plasminogen activator inhibitor-1 (PAI-1) is associated with the pathophysiology of several diseases, including cancer and cardiovascular disease. The extracellular matrix protein vitronectin increases at sites of vessel injury and is also present in fibrin clots. Integrins present on the cell surface bind to vitronectin and anchor the cell to the extracellular matrix. However, the binding of PAI-1 to vitronectin prevents this interaction, thereby decreasing both cell adhesion and migration. We previously developed PAI-1–specific RNA aptamers that bind to (or in the vicinity of) the vitronectin binding site of PAI-1. These aptamers prevented cancer cells from detaching from vitronectin in the presence of PAI-1, resulting in an increase in cell adhesion. In the current study, we used in vitro assays to investigate the effects that these aptamers have on human aortic smooth muscle cell (HASMC) and human umbilical vein endothelial cell (HUVEC) migration, adhesion, and proliferation. The PAI-1–specific aptamers (SM20 and WT15) increased attachment of HASMCs and HUVECs to vitronectin in the presence of PAI-1 in a dose-dependent manner. Whereas PAI-1 significantly inhibited cell migration through its interaction with vitronectin, both SM20 and WT15 restored cell migration. The PAI-1 vitronectin binding mutant (PAI-1AK) did not facilitate cell detachment or have an effect on cell migration. The effect on cell proliferation was minimal. Additionally, both SM20 and WT15 promoted tube formation on matrigel that was supplemented with vitronectin, thereby reversing the PAI-1's inhibition of tube formation. Collectively, results from this study show that SM20 and WT15 bind to the PAI-1's vitronectin binding site and interfere with its effect on cell migration, adhesion, and tube formation. By promoting smooth muscle and endothelial cell migration, these aptamers can potentially eliminate the adverse effects of elevated PAI-1 levels in the pathogenesis of vascular disease.
Introduction
Aptamers are single-stranded nucleic acids, either DNA or RNA, that bind to their target protein with high affinity and specificity. They are typically generated by the systematic evolution of ligands via exponential enrichment (SELEX). Adopting the SELEX in vitro selection technique ensures the development of RNA molecules that will bind to target proteins. Importantly, aptamers have the potential to block a single function of proteins without obstructing their other functions. For example, we and others (Blake et al., 2009; Madsen et al., 2010) have shown that PAI-1 RNA-specific aptamers, which bind to the PAI-1's vitronectin binding site, only affects the PAI-1–vitronectin interaction. These aptamers have been shown to have no effect on PAI-1's proteolytic activity (Blake et al., 2009; Madsen et al., 2010).
The extracellular matrix protein vitronectin increases at sites of vessel injury and is also present in fibrin clots. In response to injuries, vitronectin facilitates cell adhesion, which increases vascular cell migration by binding to integrins and surface-bound uPA (Fig. 1). The PAI-1 competes with integrins and the urokinase-type plasminogen activator receptor (uPAR) for vitronectin binding (Fig. 1), resulting in cell detachment from the extracellular matrix (Deng et al., 1996; Deng et al., 2001; Czekay et al., 2003). PAI-1's binding to vitronectin prevents integrins from binding to vitronectin while also inhibiting cell adhesion and the migration of several cell types (Fig. 1), including smooth muscle cells (SMCs) and endothelial cells (ECs) (Stefansson and Lawrence, 1996; Kjoller et al., 1997; Waltz et al., 1997; Deng et al., 2001; Stefansson et al., 2007). Our laboratory's ability to develop aptamers that bind to the vitronectin binding site of PAI-1 has made possible an investigation into the importance of the PAI-1–vitronectin interaction with regards to cell migration, adhesion, proliferation, and tube formation. Moreover, we are in a position to determine whether aptamers are able to therapeutically regulate PAI-1's adverse effect on intimal hyperplasia and tumor angiogenesis. Given that PAI-1 plays an important role in regulating cell migration, adhesion, and proliferation in vascular SMCs and ECs, we have sought to investigate the effect of our PAI-1–specific aptamers on these cellular processes.
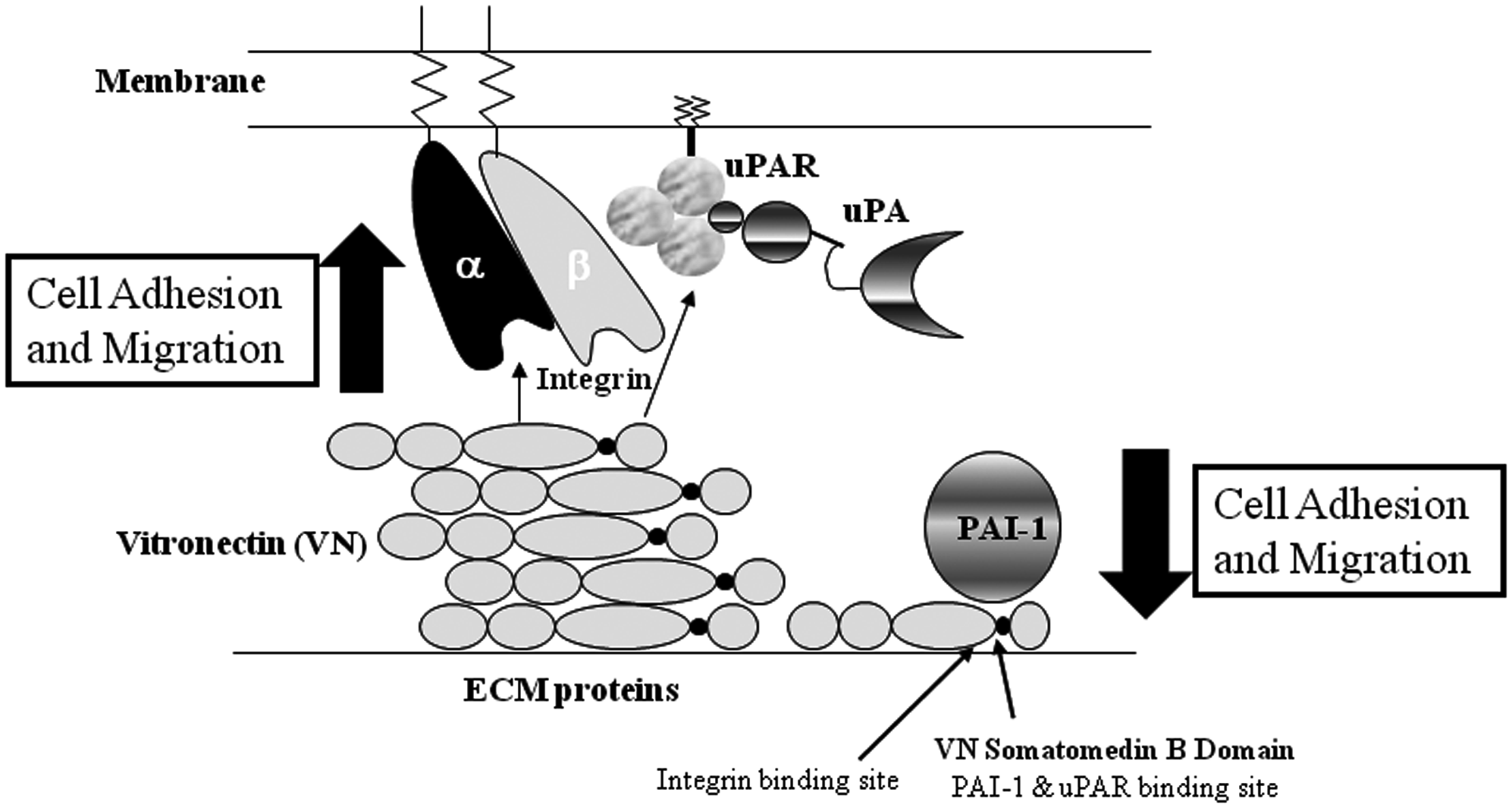
Plasminogen activator inhibitor-1 (PAI-1)–vitronectin interaction. Vitronectin expression is increased in response to vascular injury. In this state, vitronectin binds to membrane-bound integrins via the integrin binding site (arqinine glycine aspartic acid sequence) resulting in an increase in cellular adhesion and migration. Vitronectin also binds to urokinase-type plasminogen activator receptor (uPAR) (left panel). PAI-1 binds to the vitronectin's somatomedin B domain (which is also the binding site for uPAR). The integrin binding site overlaps with the somatomedin B domain. As a result, the binding of PAI-1 to vitronectin prevents the vitronectin–integrin interaction causing a decrease in cell adhesion and migration (right panel). Our PAI-1–specific RNA aptamers prevent PAI-1 from binding to vitronectin. This figure is based on a schematic from Carter and Church (2009).
Materials and Methods
Reagents
Recombinant wild-type PAI-1 (wtPAI-1; active fraction) and human monomeric vitronectin were purchased from Molecular Innovations, Inc. We purified recombinant stable PAI-1 mutant from E. coli as previously described (Whitley and Church, 2005). This mutant does not convert to the inactive form and has a half-life of greater than 140 hours. We used this stable mutant in experiments requiring longer incubation times, such as the migration assays. The aptamers used in our study (WT15 and SM20) bind with high affinity to both forms of PAI-1 (Blake et al., 2009). Recombinant PAI-1 vitronectin mutant (PAI-1AK) and stable PAI-1 mutant were generously supplied by Dr. Dan Lawrence (University of Michigan Medical School, Ann Arbor, MI). It should be noted that PAI-1AK exhibits decreased binding to vitronectin (Arroyo De Prada et al., 2002; Jensen et al., 2004). Human vitronectin and BD Matrigel™ were purchased from BD Biosciences; vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF)-B-chain homodimer glycoprotein (BB) were purchased from Invitrogen.
Cell culture
Human aortic SMCs (HASMCs), purchased from Cambrex Bio Science Walkersville, Inc., were cultured according to a standard protocol. The HASMCs were grown in SMC basal medium supplemented with 5% fetal bovine serum, human recombinant basic fibroblast growth factor (2 ng/mL), epidermal growth factor (0.5 ng/mL), heparin (5 ng/mL), insulin (5 μg/mL), and bovine serum albumin (BSA; 0.2 μg/mL). HASMCs at passages 3–8 were used in all experiments. Human umbilical vein endothelial cells (HUVECs), purchased from Invitrogen, were cultured in EC media (ScienCell Research Laboratories) supplemented with 5% fetal bovine serum and EC growth supplement (ScienCell Research Laboratories). HUVECs at passages 3–7 were used in all experiments. We maintained all cells in a humidified chamber with 5% CO2 at 37°C.
RNA aptamer in vitro transcription
We developed RNA aptamers (WT15 and SM20) as described previously (Blake et al., 2009). The cDNAs were transcribed to RNA using a DuraScribe T7 transcription kit (Epicenter Biotechnologies). Briefly, 2 μg of linearized template DNA and the T7 promoter were incubated with 100 mM DTT, 50 mM ATP, GTP, 2′-F-dCTP, and 2′F-dUTP in the presence of 10 mM Durascribe T7 enzyme mix. The reaction was incubated at 37°C for 6 hours prior to adding DNase I (1 MBU) in order to remove the DNA template. The transcript was then extracted with phenol/chloroform/isoamyl alcohol. An equal volume of 2× formamide loading buffer was then added and incubated at 65°C for 5 minutes. The RNA transcript was cooled to room temperature and subjected to electrophoresis on a 12% 7M Urea denaturing gel. The RNA was visualized by UV shadowing, excised from the gel, minced, and incubated in 2 mL TE buffer overnight at 4°C. The next day, we removed the RNA and concentrated it using Amicon Ultra centrifugal filters (Millipore). The RNA concentration was determined and then used in subsequent experiments. The RNA aptamers were incubated at 65–75°C for 5 minutes before being used in all experiments.
Cell migration assays
We performed migration assays using BD culture inserts (BD Biosciences) coated with vitronectin (5 μg/mL) containing an 8-μm-diameter pore size membrane. Vitronectin-coated membranes were hydrated with serum-free medium containing 0.1% BSA for 2 hours at 37°C prior to the experiments. The chemoattractant, serum-free medium containing either VEGF (1 ng/mL; HUVECs) or PDGF (10 ng/mL; HASMCs), was added to the lower well (750 μL) of the plate. Coated inserts were treated with PAI-1 (100 nM) or PAI-1 (100 nM) preincubated with aptamers (200–500 nM) 30 minutes prior to adding cells. Approximately 5×104 cells were seeded in the upper well (culture insert) in serum-free medium, with or without treatment, as indicated and incubated for 18–24 hours at 37°C. Following the incubation, the media were removed from the insert and cells on the upper surface of the insert were removed using a cotton-tipped applicator. The migrated cells located on the lower surface of the membranes were fixed with 70% ethanol for 5 minutes. Inserts were then washed with either distilled H2O or 1×phosphate-buffered saline (PBS) and stained with Hematoxylin Gill's Formula (Vector Laboratories, Inc.) for 5 minutes. The membranes were excised from the insert, inverted, and mounted on glass microscope slides. The total number of cells was counted in four fields at 10×magnification using light microscopy.
Cell adhesion assays
Cell adhesion assays were performed as previously described (Palmieri et al., 2002). Wild-type PAI-1 (100 nM), with or without aptamers (WT15 or SM20; 100–200 nM), was added to 96-well culture plates coated with vitronectin (7.5 μg/mL) and incubated at 37°C for 30 minutes. Aptamers were incubated with PAI-1 for 10 minutes at room temperature before being added to plates. Afterward, either HASMCs or HUVECs (5×104) were added and subsequently incubated at 37°C for 1–2 hours in serum-free media. After the incubation period, the nonadherent cells were gently removed with a multichannel pipette and the plate was washed twice with 1×PBS. The number of attached cells was determined by performing an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Briefly, 10 μg of MTT solution (5 mg/mL) was added to each well containing 100 μL of serum-free media and incubated at 37°C for 2 hours. After the incubation period, the media were aspirated and the MTT crystals were dissolved in dimethyl sulfoxide. The absorbance was measured using a VersaMax microtiter plate (Molecular Devices) reader at 600 nm.
Cell proliferation assays
HUVECs or HASMCs (2×103 cells/well) were plated in 96-well cultured plates in basal medium containing 0.5% fetal bovine serum. The cells were incubated overnight at 37°C. The next day, PAI-1 (50–100 nM), with or without aptamers (100–200 nM), was added to the plate and incubated for 24–72 hours at 37°C. PAI-1 was incubated with aptamers for 10 minutes prior to adding to cells. To determine cell proliferation, a CyQuant NF cell proliferation assay kit (Invitrogen/Molecular Probes) was used according to the manufacturer's protocol.
Binding assay
Conditioned media from HUVECs treated with PAI-1 (100 nM), PAI-1/SM20 complexes, or SM20, were collected after 24 hours. Binding of SM20 RNA aptamer to secreted proteins present in the conditioned media was determined by nitrocellulose filter binding assays as described previously (Rusconi et al., 2000). Briefly, RNA was dephosphorylated using bacterial alkaline phosphatase (Gibco BRL) and 5′ end labeled with [γ-32P] ATP (PerkinElmer Life and Analytical Sciences, Inc.) using T4 polynucleotide kinase (New England Biolabs). Conditioned media were incubated with labeled RNA for 15 minutes prior to being added to the nitrocellulose membrane. The membranes were washed and the RNA bound to the membranes was determined by counting radioactivity using a liquid scintillation counter (Beckman Coulter, Inc.).
In vitro tube formation assays
This assay was used to assess angiogenesis in vitro by evaluating the ability of ECs to form capillary-like structures (tubes) when plated on a suitable extracellular matrix support. Matrigel (BD Biosciences) was added to the wells of a treated 15-well μ-slide angiogenesis microscope slide (ibidi) in a volume of 10 μL and allowed to solidify at 37°C for 30 min. After the Matrigel solidified, HUVECs (1.5×104 cells) were added in 50 μL of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. The cells were incubated at 37°C with humidified 95% air/5% CO2 for 18 hours in the presence or absence of PAI-1 (50–200 nM) or PAI-1/aptamer (200–500 nM). The tubes (cells) were then labeled with Calcein AM Fluorescent Dye (8 μg/mL; BD Biosciences, San Jose, CA) for 30–45 minutes at 37°C, 5% CO2, viewed using a fluorescent microscope (Olympus BX61), and photographed. The number of tubes was counted in four independent fields at 4×magnification.
Statistical analysis
Statistical analysis was performed using InStat (Graph Pad Software, Inc.). A one-way analysis of variance test was used followed by a Dunnett multiple comparison tests. A P value less than 0.05 was considered significant with a 95% confidence level (the differences between the means were true).
Results
Cell adhesion to vitronectin is regulated by SM20 and WT15
Previously, we reported that the PAI-1–specific RNA aptamer, SM20, and to a lesser extent WT15, restored cellular adhesion of the breast cancer cell line, MDA-MB-231, to vitronectin in the presence of PAI-1 (Blake et al., 2009). In the current study, we investigated whether SM20 and WT15 are also able to alter the adhesion of SMCs and ECs to immobilized vitronectin when exposed to PAI-1. The data in Table 1 represent percent cell adhesion normalized to the adherence of cells to vitronectin in the absence of PAI-1, which was set at 100%. Both PAI-1 and the PAI-1–aptamer complexes, respectively, were added to immobilized vitronectin prior to adding the cells. PAI-1 inhibited the attachment of both HASMCs and HUVECs to vitronectin (Table 1) by approximately 30%. These results are in agreement with the literature (Leik et al., 2006). No significant effect was observed when PAI-1 was incubated with the control aptamer library, Sel2, which does not bind with high affinity to PAI-1 (Blake et al., 2009). When PAI-1 was preincubated with either SM20 or WT15, we observed a concentration-dependent increase in cell attachment of HASMCs and HUVECs to immobilized vitronectin (Table 1). The PAI-1 vitronectin mutant (PAI-1AK) did not have a significant effect on the attachment of either cell to vitronectin (data not shown). HASMCs and HUVECs attached normally when incubated with aptamers alone, indicating that both SM20 and WT15 bind specifically to PAI-1 and not to vitronectin (Table 1). These results demonstrate that SM20 and WT15 promote the adhesion of SMCs and ECs to vitronectin in the presence of PAI-1. This further supports our findings that SM20 and WT15 are able to effectively prevent PAI-1 from binding to vitronectin and are capable of regulating cell adhesion.
Data are expressed as percentage of adhesion of SMCs and endothelial cells to vitronectin. All data are normalized to adhesion of cells to vitronectin in the absence of PAI-1. PAI-1 was used at a concentration of 100 nM.
HASMCs, human aortic smooth muscle cells; HUVECs, human umbilical vein endothelial cells.
SM20 and WT15 increase migration of SMCs and ECs
PAI-1 has been shown to block SMC (Schneider et al., 2004) and EC migration (Deng et al., 1996; Kjoller et al., 1997); this interaction is partly credited to the PAI-1–vitronectin interaction (Stefansson et al., 2007). Taking this into consideration, we sought to determine whether SM20 and WT15 regulate the motility of HASMCs and HUVECs. Cells were plated onto transwell membranes coated with vitronectin and stimulated with VEGF (HUVECs) or PDGF-BB (SMCs). As expected, PAI-1 decreased the migration of both SMCs and HUVECs (Fig. 2). In contrast, when the cells were treated with either PAI-1/SM20 or PAI-1/WT15 complexes, cell migration increased in comparison to being treated with wtPAI-1 alone (Fig. 2). When PAI-1 was incubated with the control aptamer library (Sel2), only a minor increase in cell migration could be detected (Fig. 2). At higher concentration of Sel2 (500 nM), no significant increase in cell migration was detected (data not shown). WT15 generated a greater effect on cell migration as compared with SM20; however, this difference was not significant. Adding WT15 and SM20 aptamers alone slightly increased the migration of HASMCs and HUVECs, which is most likely due to the aptamers binding to secreted PAI-1 (data not shown).
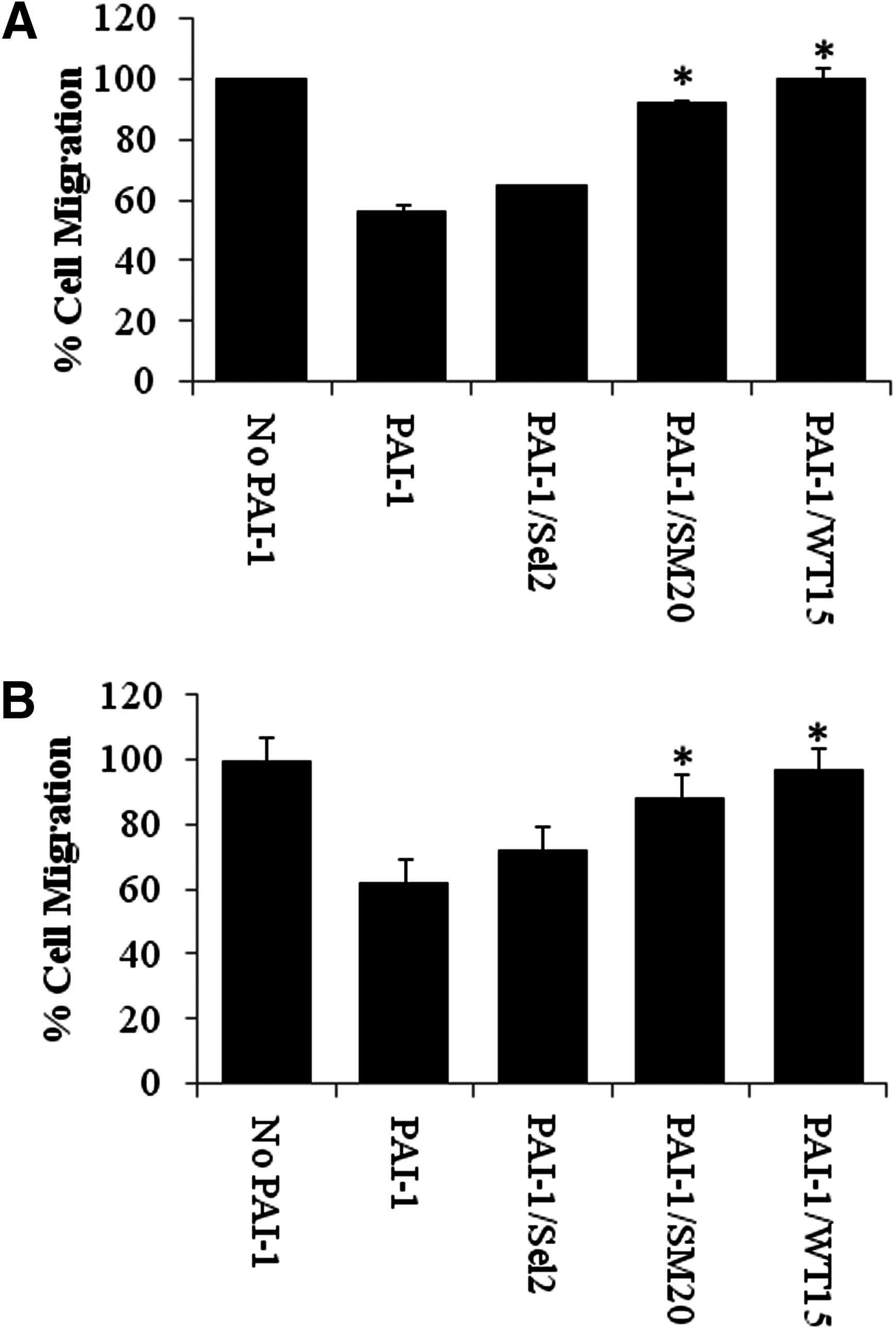
Effects of PAI-1 and PAI-1–specific RNA aptamers on the migration of SMCs and endothelial cells. Wild-type PAI-1 (100 nM) alone, PAI-1 in the presence of the PAI-1–specific aptamers (200 nM), or PAI-1 in the presence of the control aptamer, Sel2 (200 nM), were added to vitronectin-coated transwell inserts for 30 minutes before adding HASMCs
The PAI-1–vitronectin interaction has a minor effect on cell proliferation
In vitro assays were conducted to determine how PAI-1 and PAI-1 incubated with WT15 and SM20 impacted cell proliferation. PAI-1 inhibited the proliferation of HASMCs and HUVECs in the absence of aptamers. Both WT15 and SM20 slightly increased the proliferation of HASMCs in the presence of PAI-1 (Fig. 3A). Surprisingly, WT15 and SM20 caused a slight decrease in HUVEC proliferation when compared with cells treated with PAI-1 alone (Fig. 3B). Nevertheless, neither aptamer bore a significant effect (increase or decrease) on the proliferation of either cell line (Fig. 3).
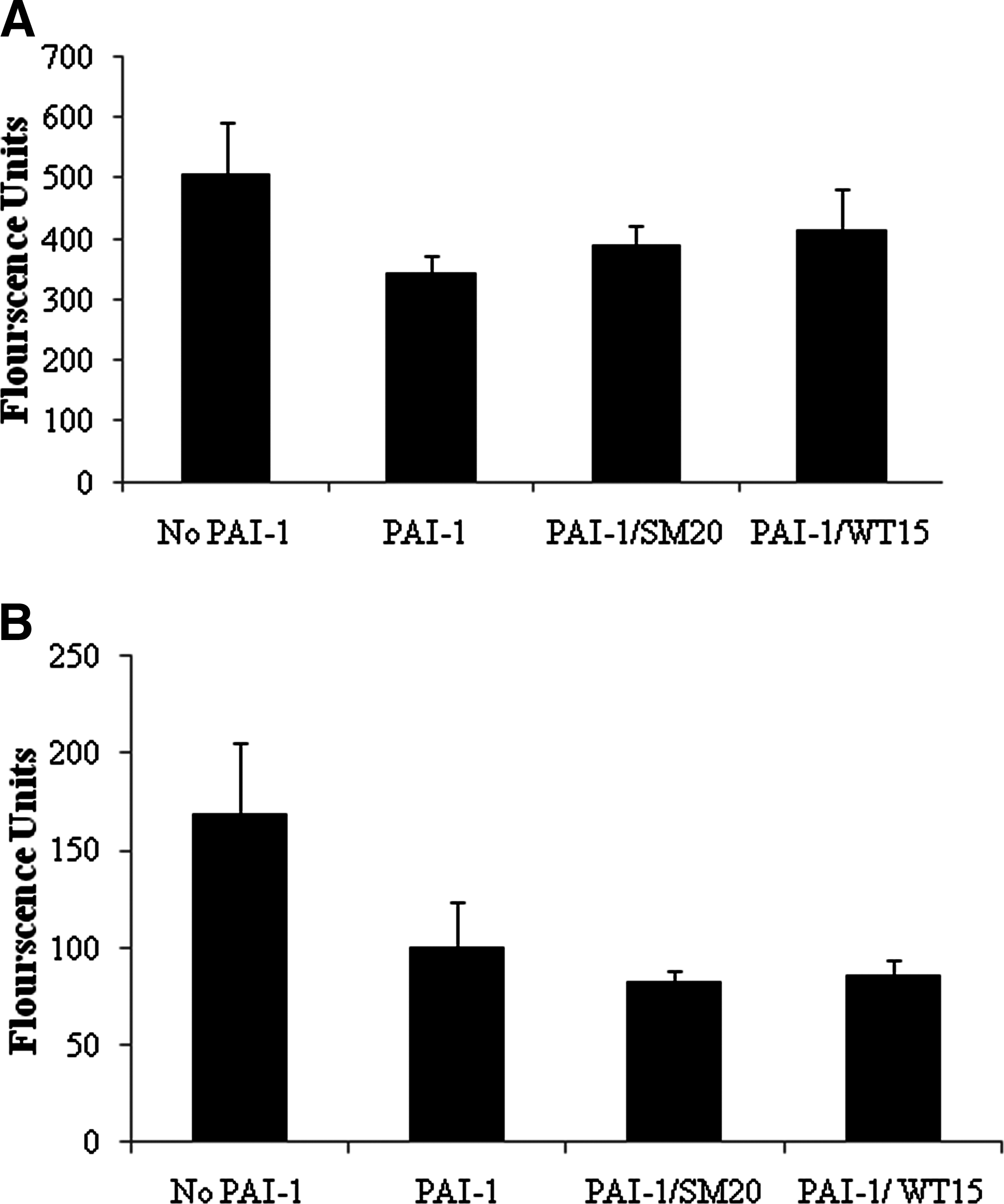
Effects of PAI-1 and PAI-1–specific aptamers on proliferation of SMCs and endothelial cells. HASMCs
The aptamers bind to secreted PAI-1
Next, we wanted to determine whether the aptamers bind to PAI-1 secreted from HUVECs. For this experiment, we end-labeled SM20 RNA with 32P and incubated it with conditioned media from HUVECs. Binding was gauged using the nitrocellulose binding assay (as described in Materials and Methods). Approximately 25% of the labeled aptamer effectively bound to secreted proteins in the absence of PAI-1. However, when PAI-1 was added, we observed an increase in binding (Fig. 4A). Subsequently, when the cells were incubated with SM20 alone aptamer binding decreased. The most plausible explanation for this observation is that “cold” (unlabeled) SM20 was bound to PAI-1, thereby preventing “hot” (radioactively labeled) SM20 from efficiently binding.
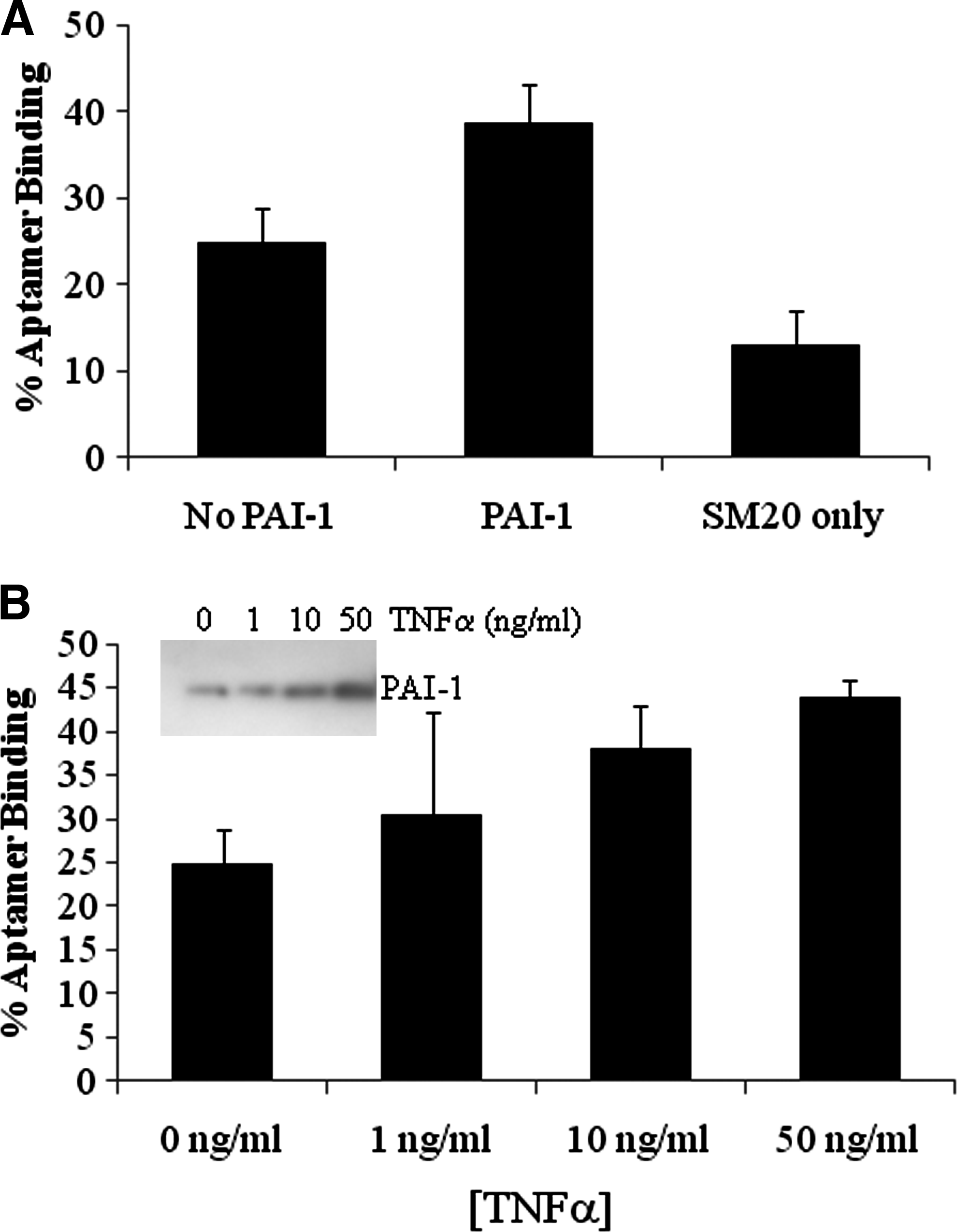
PAI-1–specific RNA aptamers bind to secreted and exogenous PAI-1. The HUVECs (1×106) were seeded in six-well culture dishes in the presence of PAI-1, the PAI-1/SM20 complex, or SM20 alone, overnight. The binding of 32P-labeled SM20 aptamer to secreted proteins was assessed by nitrocellulose binding assay as described in Materials and Methods
It is possible that our aptamers could bind to other proteins secreted into the conditioned medium. Taking this into account, we further investigated SM20's ability to bind PAI-1 that was secreted by cells following induction by TNFα. TNFα increases the production of PAI-1 in a concentration-dependent manner in endothelial cells and SMCs (van Hinsbergh et al., 1988; Gallicchio et al., 1994). An increase in TNFα concentration correlates with both an increase in PAI-1 protein (Fig. 4B inset) and aptamer binding (Fig. 4B). Similar results were obtained with the WT15 aptamer (data not shown). Collectively, these results imply that SM20 binds to both endogenous (secreted) and exogenous PAI-1.
Effect of PAI-1–specific RNA aptamers on EC tube formation
To determine whether WT15 and SM20 were capable of altering PAI-1–mediated inhibition of angiogenesis, we examined SM20's and WT15's effect on the formation of tubes by HUVECs that were plated on matrigel supplemented with vitronectin. Cells treated with PAI-1 (100 nM) formed significantly fewer tubes compared with untreated cells (Fig. 5A). This corroborates previous studies (Isogai et al., 2001). Incubating PAI-1 with either WT15 or SM20 increased the number of tubes formed (Fig. 5B). In addition, incubating the cells with PAI-1AK resulted in the formation of significantly more tubes. The control aptamer library had no effect on this process (data not shown). Incubating aptamers in the absence of PAI-1 did not alter the number of tubes formed or the morphology of the tubes (data not shown); such evidence further supports our hypothesis that WT15 and SM20 can regulate PAI-1's role in angiogenesis by disrupting its interaction with vitronectin.
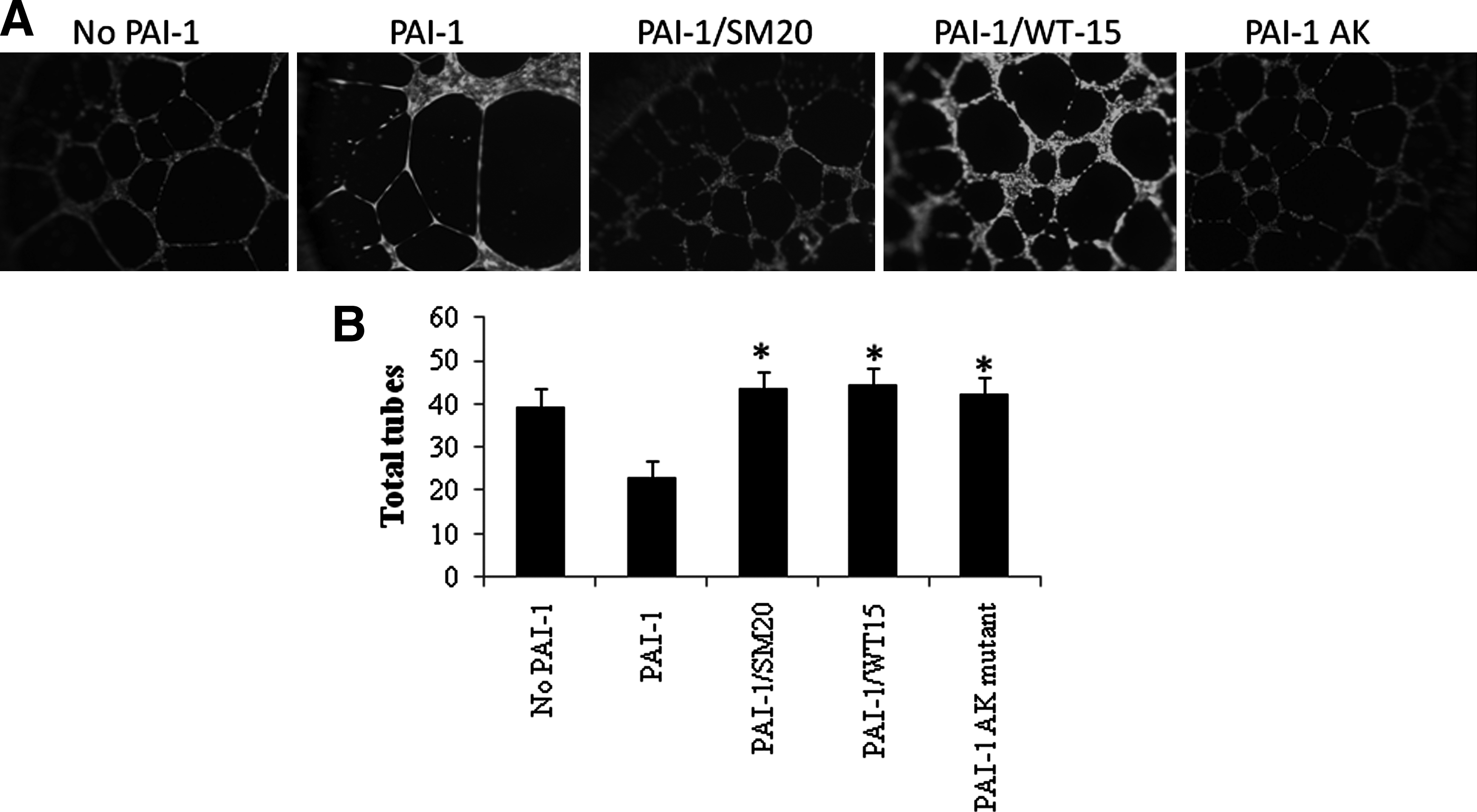
Effects of PAI-1 and PAI-1–specific aptamers on tube formation. The HUVECs (1.5×104) were placed on matrigel supplemented with vitronectin (5 mg/mL). PAI-1, PAI-1 preincubated with aptamers, or the PAI-1AK mutant, was added to the cells and incubated at 37°C. Tubes formed within 24 hours. The slides were photographed
Discussion
Developing novel PAI-1 inhibitors is the focus of several research groups (Crandall et al., 2003; Izuhara et al., 2008; Miyazaki et al., 2008; Suzuki et al., 2008; Izuhara et al., 2010). These agents are designed to regulate PAI-1's adverse activity (for review see Suzuki et al., 2011). As a result, several of them are chemical suppressors that have shown promise in both in vivo and clinical studies (Elokdah et al., 2004; Suzuki et al., 2008; Izuhara et al., 2010; Ogawa et al., 2011). Many of the PAI-1 inhibitors, such as IMD-1622 and TM5001, disrupt the interaction of PAI-1 with tPA, disturbing its proteolytic activity (Suzuki et al., 2008; Izuhara et al., 2010). The interaction of PAI-1 with vitronectin is also important in generating PAI-1's adverse effects, particularly with respect to regulating cell adhesion, proliferation, and migration. When designing PAI-1 inhibitors, one must take into consideration that PAI-1 is a multifunctional protein containing several domains, each playing a critical role in pathophysiological events. Consequently, although not our initial objective, we developed molecules that interfere with one domain of PAI-1 without disrupting the function of the other domains by using RNA aptamers. This allows us to analyze each domain individually. Our previous studies revealed that disrupting the PAI-1's binding to vitronectin with RNA aptamers brought increases in the adhesion of breast cancer cells to vitronectin (Blake et al., 2009). Importantly, we also showed that these aptamers did not alter PAI-1's ability to interact with tPA (Blake et al., 2009). However, in this study, we expanded upon our initial findings to investigate whether our PAI-1–specific aptamers could regulate cell adhesion, migration, and proliferation of SMCs and ECs. Both cell types are vital for both tissue remodeling and angiogenesis and are regulated effectively by PAI-1. We demonstrated that two PAI-1–specific RNA aptamers (SM20 and WT15) restored SMC/EC adhesion and migration without significantly affecting cell proliferation. We also showed how the PAI-1–induced inhibition of EC tube formation in the presence of vitronectin is partially reversed by our aptamers. Collectively, our findings underscore the potential of aptamers to serve as therapeutic molecules for regulating PAI-1's activity at the cellular level.
Several inhibitory aptamers are currently in clinical trials or are being used to treat certain diseases (Ruckman et al., 1998; Rusconi et al., 2002; Hicke et al., 2006; Ng et al., 2006; Gilbert et al., 2007; Bates et al., 2009). For example, the VEGF-specific RNA aptamer was approved by the Food and Drug Administration (2004) for the treatment of age-related macular degeneration (Ng et al., 2006). Numerous inhibitory aptamers that target blood clotting factors, particularly thrombin and factor IXa, are in various phases of clinical trials and are yielding promising results (Rusconi et al., 2002; Muller et al., 2007; Oney et al., 2007). Hence, aptamers bear potential for therapeutically regulating PAI-1's activity. To our knowledge, there is only one other group that has developed inhibitory aptamers to PAI-1 (Madsen et al., 2010). Madsen et al. (2010) reported the generation of two PAI-1 aptamers that, like ours, affected PAI-1's binding to vitronectin. This group demonstrated that their aptamers do not affect PAI-1's antiproteolytic activity (Madsen et al., 2010). Their findings agree with our research (Blake et al., 2009). Interestingly, it is important to note that there are differences between our aptamer sequences and those of Madsen et al. (2010). The divergence might be attributed to the respective origins of our purified proteins. Additionally, our aptamers may span across different, but overlapping, PAI-1 regions, with both having an effect on the interaction of PAI-1 with vitronectin. Although we have not elucidated the exact binding site of our aptamers, we are confident that they bind to an area encompassing the heparin/vitronectin binding site (Blake et al., 2009).
PAI-1's binding to vitronectin inhibits the adhesion and migration of vascular SMCs and ECs by blocking the interaction of integrins with vitronectin (Stefansson and Lawrence, 1996; Kjoller et al., 1997; Stefansson et al., 2007). PAI-1 has also been shown to promote SMC migration by binding to the uPA–uPAR complex (Degryse et al., 2004; Whitley and Church, 2005). When PAI-1 is attached to uPA–uPAR, PAI-1 reverts to a conformation that is favorable for high-affinity binding to the LDL receptor–related protein (Stefansson et al., 1998; Czekay and Loskutoff, 2004; Degryse et al., 2004). This internalizes the uPAI–PAI-1–uPA complex, which permits SMCs to detach itself from the extracellular matrix and promote migration. Such an effect on migration is independent of vitronectin binding (Degryse et al., 2004; Whitley and Church, 2005). Disrupting the PAI-1–vitronectin interaction increases both adhesion and migration, thereby confirming previous research (Leik et al., 2006; Stefansson et al., 2007). Both SM20 and WT15 equally augmented the adhesion of SMCs and ECs to vitronectin. Similar effects on the migration and adhesion of SMCs were found using the PAI-1 inhibitor, PAI-039 (Leik et al., 2006). The one principal difference, however, involved variances in the concentration of drug/RNA required to produce an effect. Unlike PAI-039, our aptamers elicited responses at nanomolar, as opposed to micromolar, concentrations (Leik et al., 2006). This is not surprising given the high-affinity binding of aptamers to their target proteins. Another difference is that PAI-039 also inhibits PAI-1's antiproteolytic activity (Leik et al., 2006), whereas neither SM20 nor WT15 affects this process.
Meanwhile, another study examining the effects of treating SMCs (isolated from wild-type mice) both with active stable PAI-1 and a PAI-1 mutant lacking antiproteolytic activity demonstrated that both molecules decrease cell migration (Wu et al., 2009). When these cells were treated with the PAI-1AK mutant, cell migration remained unaltered, confirming that PAI-1–mediated decreases in cell migration are due to PAI-1's binding to vitronectin (Wu et al., 2009). Similarly, both SM20 and WT15 effectively prevented PAI-1 from inhibiting SMCs from migrating. This demonstrates the importance of the PAI-1–vitronectin interaction in the migration of SMCs. Consequently, controlling the migration of SMCs is critical for regulating several diseases (such as atherosclerosis), as inhibition of SMC migration can produce unstable plaques that are prone to rupture.
PAI-1 both inhibits (Ploplis et al., 2004; Balsara et al., 2006; Wu et al., 2009) and promotes cell proliferation (Chen et al., 2006). Regulating vascular SMC proliferation by means of PAI-1 is vital in vascular remodeling, especially since overexpressing PAI-1 promotes intimal hyperplasia, which negatively impacts vascular remodeling (Kauhanen et al., 1997). Though PAI-1 did not inhibit the proliferation of vascular SMCs when isolated from vitronectin knockout mice, PAI-1 did successfully inhibit the proliferation of SMCs from wild-type mice (Wu et al., 2009). In our proliferation assays, we showed that PAI-1 inhibits the proliferation of both SMCs and ECs, a finding that agrees with the extant literature (Wu et al., 2009). Neither WT15 nor SM20 significantly augmented PAI-1–mediated decreases in the proliferation of either cell type. These results may initially seem perplexing. However, Wu et al. (2009) concluded that not only did wtPAI-1 inhibit cell proliferation, but the PAI-1AK mutant, which lacks the ability to bind to vitronectin, also inhibited the proliferation of SMCs when isolated from wild-type mice. Consequently, the simple interaction of PAI-1 with endogenously expressed vitronectin does not solely account for its role in cell proliferation. Indeed, these authors suggest that the antiproliferative effects of PAI-1AK on vascular SMCs are potentially attributable to the inhibition of cell-associated plasminogen activators or other proteases, such as thrombin (Wu et al., 2009). Since PAI-1's antiproteolytic activity is reserved when bound to our aptamers (Blake et al., 2009), it is also possible that the antiproliferative effect that we observed is equally due to protease inhibition. Also possible is that neither WT15 nor SM20, at the concentrations used in this experiment, was able to displace vitronectin bound to PAI-1. Although we have revealed that both SM20 and WT15 compete with vitronectin for binding to PAI-1 (Blake et al., 2009), we must underscore that our findings were achieved in solution, not on cells. Equally, it is possible that SM20 and WT15 are unable to inhibit PAI-1 once bound to vitronectin, as is the case with the PAI-039 inhibitor (Leik et al., 2006). More research is needed to adequately address these issues.
It is widely accepted that PAI-1 plays a role in regulating angiogenesis. Yet, as with cell migration, adhesion, and proliferation, PAI-1 has been shown to both promote and inhibit (Isogai et al., 2001; Stefansson et al., 2001) angiogenesis. One factor that is important for PAI-1–mediated regulation of angiogenesis involves the composition of the extracellular matrix. Isogai et al. (2001) established that PAI-1 inhibits EC tube formation on matrigel that is supplemented with vitronectin. However, it was shown that fibronectin actually stimulated tube formation (Isogai et al., 2001). In addition, others have asserted that the binding of PAI-1 to vitronectin can be attributed to the inhibition of VEGF-induced angiogenesis (as seen in the chicken chorioallantoid membrane assays) (Stefansson et al., 2001). We demonstrate that PAI-1 effectively inhibits EC tube formation on matrigel that is supplemented with vitronectin, a finding that concurs with the general literature (Isogai et al., 2001). Both WT15 and SM20 were able to abate the PAI-1–mediated inhibition, implying that the PAI-1–vitronectin interaction is critical for the inhibition of angiogenesis. Interestingly, the PAI-1 inhibitor PAI-039, which also disrupts binding of PAI-1 to vitronectin, inhibited angiogenesis in vivo. Our studies, however, were performed in vitro; therefore, additional in vivo research is needed to further compare the two inhibitors. Moreover, PAI-039 not only prevents PAI-1 from interacting with vitronectin, but also from inhibiting PAI-1 that is bound to tPA (Leik et al., 2006). Consequently, the PAI-1–mediated effect on angiogenesis may be both vitronectin and protease dependent. Nevertheless, collectively, previous research and our results further indicate that the vascular environment is critical toward establishing the overall PAI-1–mediated angiogenic effect.
Footnotes
Acknowledgments
The authors would like to thank Dr. Daniel Lawrence for his generous donation of PAI-1 vitronectin mutant and the human stable PAI-1 mutant. This work was supported by National Institutes of Health (NIH) grant #HL096407 awarded to Yolanda M. Fortenberry and #HL65222 awarded to Bruce Sullenger.
Disclosure Statement
No competing financial interests exist.