
Abstract
Avian influenza is an acute viral respiratory disease caused by RNA viruses of the family Orthomyxoviridae. The influenza A virus subtype H5 can cause severe illness and results in almost 100% mortality rate among livestock. Hemagglutinin (HA) present in the virus envelope plays an essential role in the initiation of viral infection. In this study, we investigated the efficacy of using HA as a target for antiviral therapy through nucleic acid aptamers. After purification of the receptor binding domain (HA1) of HA protein, activity of recombinant HA1 was confirmed by using hemagglutination assay. We selected RNA aptamer candidates after 15 rounds of iterative Systematic Evolution of Ligands by EXponential enrichment (SELEX) targeting the biologically active HA protein. The selected RNA aptamer HAS15-5, which specifically binds to HA1, exhibited significant antiviral efficacy according to the results of a hemagglutination inhibition assay using egg allantoic fluids harboring the virus. Thus, the RNA aptamer HAS15-5, which acts by blocking and inhibiting the receptor-binding domain of viral HA, can be developed as a novel antiviral agent against type H5 avian influenza virus.
Introduction
HA binds to lung epithelial cell receptors that contain sialic acid, which subsequently allows the virus to enter the host cell through the endosomal pathway (Skehel and Wiley, 2000; Eckert and Kim, 2001). HA is synthesized as a precursor protein (HA0) and has to be cleaved by a protease into the HA1 and HA2 subunits (Klenk et al., 1975; STEINHAUER, 1999). HA is a primary target for antiviral agents, and the host cell receptor-binding site in the HA1 subunit has been shown to be a viable antiviral target site (Jeon and Arnon. 2002; Jeon et al., 2004). Furthermore, the truncated recombinant HA protein containing the HA1 region has been shown to induce an immune response against the influenza virus in humans (Jeon and Arnon, 2002). Several small molecular inhibitors against HA of influenza A virus, such as CL-385319 (Plotch et al., 1999), stachyflin (Yoshimoto et al., 2000), and BMY-27709 (Luo et al., 1997), have been developed and investigated for the mechanism of action against H5N1 influenza A virus (Liu et al., 2011).
Aptamers are oligonucleotides or peptide molecules that bind to a specific target molecule and are created using an in vitro selection process called the Systematic Evolution of Ligands by EXponential enrichment (SELEX) (Ellington and Szostak, 1990; Tuerk and Gold, 1990). Aptamers have many advantages over antibodies, such as high affinity, low costs, low-temperature sensitivity, large-scale production, long shelf-life, and reversible denaturation (Nimjee et al., 2005). Due to these remarkable properties, nucleic acid aptamers have been recently used as biosensors or therapeutic tools against virus and cancer (Perkins and Missailidis, 2007; Tombelli and Mascini, 2009). Several RNA aptamers have been selected as antiviral agents because they can target and inhibit viral proteins such as severe acute respiratory syndrome (SARS), coronavirus helicase (Jang et al. 2008), human immunodeficiency virus (HIV) Tat (Yamamoto et al., 2000), reverse transcriptase (Tuerk et al., 1992), hepatitis C virus (HCV) NS3 protease (Fukuda et al., 2000), helicase (Hwang et al., 2004; Umehara et al., 2005), and NS5B RNA-dependent RNA polymerase (Biroccio et al., 2002). Recently, DNA aptamers targeting HA of H5N1 influenza virus have been selected and tested for antiviral activity (Cheng et al., 2008).
In the present study, we performed a SELEX procedure for the isolation of RNA aptamers that could bind to and inhibit the recombinant HA1 protein of H5 subtype influenza virus A. We demonstrated that the resultant isolated RNA aptamer was capable of inhibiting the hemagglutination activity of viral fluids obtained from specific-pathogen-free (SPF) eggs inoculated with the H5 type influenza virus. We hope that the selected RNA aptamer, combined with appropriate therapeutic formulation, can be used as an antiviral reagent against type H5 influenza virus, through blocking the receptor-binding domain of HA.
Materials and Methods
Expression and purification of recombinant HA1 protein
Influenza virus strain A/wild-duck/Korea/ES/2004 (H5N2) was cultivated in the allantonic cavities of 11-day-old embryonated chicken eggs incubated at 37°C. The allantonic fluid was harvested 3 days after virus inoculation and used for RNA extraction and complementary DNA (cDNA) synthesis. PCR amplification of the HA1 gene (i.e., the region between the 49th and 1023rd nucleotides of the full-length HA gene) was performed with a forward primer (5′-GATAATACGACTCACTATAGGGTTCACTGCAGACTTGACGAA-3′) and a reverse primer (5′-GAATTCGTAGATGTGGATCCATT-3′). The amplified products (975 bp) of digestion with EcoRI and XhoI were cloned into the pGEX-4T-1 vector. The recombinant HA1 protein, with a glutathione-S-transferase (GST) tag at the N-terminus, was expressed by the Escherichia coli Rosetta strain, which had been cultured overnight in Luria-Bertani medium containing 100 μg/mL ampicillin at 37°C. The resulting culture (10 mL) was transferred to 1 L of LB medium containing 100 μg/mL ampicillin and was incubated in a shaker at 37°C until the optical density (OD) 600 reached 0.8. Isopropyl-β-
The GST-fused HA1 that was bound to the DEAE-Sephadex was loaded onto a Glutathione-Sepharose 4B column (GE Healthcare) pre-equilibrated with lysis buffer. The column was washed twice with 500 mL of the same buffer, and the recombinant HA1 was then eluted with an elution buffer containing 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 1% (v/v) Triton X-100, 150 mM NaCl, and 10 mM glutathione. The eluted fractions were analyzed on a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) gel to measure the presence of GST-fused HA1 protein (62.5 kDa). Pure protein fractions were pooled and dialyzed against the dialysis buffer [20 mM Tris-HCl, pH 7.5, 1.0 mM EDTA, 5 mM DTT, 20% glycerol, 0.5% (v/v) Triton X-100, and 150 mM NaCl]. Proteins were concentrated with a Centricon Plus-20 centrifugal filter (Millipore), and the concentration of the purified protein was determined by Bradford assay with bovine serum albumin (BSA) as a standard. Identity of the purified recombinant HA1 protein was confirmed by immunoblotting with a rabbit polyclonal antibody against H5 raised from H5N1 (Abcam, Cambridge, UK), followed by detection with enhanced chemiluminescence (Daeil Lab service, Seoul, Korea).
In vitro selection of RNA aptamers against HA1 protein
For the SELEX process, a pool of random sequence RNAs (RNA library) was produced by PCR and in vitro transcription of the DNA template containing 40 random nucleotides, as described previously (Jang et al., 2008). The GST-tagged HA1 protein was immobilized using Glutathione-Sepharose 4B MicroSpin columns (GE Healthcare). First, negative selection was performed to remove RNAs bound to the glutathione-sepharose resin, which was charged with separately prepared GST protein, from the RNA pool. The column was washed with PBS buffer, pH 7.4, containing 140 mM NaCl and 2.7 mM KCl. Then 6 μg of RNA library was passed through the column. Before use, the RNA library had been denatured at 85°C for 3 min and folded at room temperature in 300 μL of PBS buffer. The unbound RNA pool was collected and reloaded onto a new MicroSpin column that was charged with 50 μg of the GST-tagged HA1. The column was washed three times with 500 μL of PBS buffer, and RNAs bound to the target protein were eluted with glutathione buffer (10 mM glutathione, 50 mM TrisHCl, pH 8.0). The RNAs were purified by phenol-chloroform extraction and subsequent ethanol precipitation. The purified RNA was subjected to RT-PCR and in vitro transcription to generate an RNA pool for the next round of SELEX.
HA1 protein binding assay
To analyze the binding affinity of the 15th round RNA pool, we conducted semiquantitative reverse transcriptase polymerase chain reaction (RT-PCR) of the 15th round pool along with the RNA pools from other rounds (the initial RNA pool and the 7th round pool) and some irrelevant sequences of RNA, which was used as negative control. Specifically, 250 ng of HA1 protein was loaded and immobilized onto a Glutathione-Sepharose MicroSpin column, and 1 μg of each RNA pool was loaded onto the column, as performed in the previous SELEX processes. The column containing the RNA–protein complex was then washed three times with 200 μL of the binding buffer, after which the eluants were collected as flowthrough. The RNAs bound to HA1 protein were then eluted from the column by three consecutive washes with 200 μL of the elution buffer. RNAs from the flowthrough and the protein-bound RNAs were then extracted by phenol-chloroform and ethanol precipitation. The extracted RNAs from each elution were subjected to RT-PCR using the aptamer-specific primers described above. The conditions for the PCR were as follows: 13 cycles of 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds. Electrophoresis was conducted on 1% agarose gel to measure the amount of cDNA that had been amplified. The amplified cDNA bands on each gel were quantified using Gel-Pro Analyzer software (Media Cybernetics, Bethesda, MD).
Identification of aptamer sequences and secondary structures
After the 15th round of the SELEX process, the amplified cDNA was cloned into a linearized pGEM-T Easy vector (Promega, Madison, WI). The cloned gene was transfected into Escherichia coli, and isolated plasmid DNA from each clone was sequenced. Each RNA aptamer was generated by in vitro transcription using T7 RNA polymerase. The amplified cDNA or respective subcloned plasmid was used as a template. The secondary structures of RNA aptamer candidates were predicted by the Mfold program, which is based on the Zuker algorithm (ZUKER, 1989).
Hemagglutination assay
Recombinant HA1 and BSA that were serially diluted (10 μg to 0.005 μg) with PBS buffer (pH 7.4, 0.14 M NaCl, 2.7 mM KCl) and then were placed in a round-bottomed 96-well plate (SPL, Seoul, Korea). A mixture consisting of 40 μL of 1% (v/v) chicken red blood cells (RBCs) in PBS was then added to the each well. The reaction mixture was incubated at room temperature for 2 hours, after which the plate was photographed. For RNA aptamer selection based on anti-hemagglutination activity, serially diluted HA1 protein (1.2 μg to 0.6 μg) was mixed with 1 μg of each RNA aptamer candidate in separate wells of a round-bottomed 96-well plate. After 15 minutes of incubation at room temperature, 0.2 mL of 1% (v/v) chicken RBCs were added to each well and further incubated for 1 hour at room temperature. The plate was then photographed to assess the hemagglutination activity in each well.
Inhibition of influenza virus activity by RNA aptamer
Avian influenza H5N2 (WB/A-G63/05) virus was cultured in the allantoic sacs of 11-day-old chicken embryos at 37°C for 2 days. The allantoic fluid was harvested and stored at −70°C until use. The titer of virus in the allantoic fluid was determined as 50% of the egg-infecting dose (EID50). For inoculation, 200 μL of serially diluted allantoic fluid containing the virus and 10 μg of designated RNAs were mixed and incubated at room temperature for 30 minutes. The virus–RNA mixture was injected into the embryonated SPF eggs and incubated for 3 days at 37°C. Virus fluid was withdrawn from the SPF eggs, and the hemagglutination activity of the virus fluid was determined using chicken RBCs as described above.
Results
Preparation of biologically active HA protein for SELEX
GST-fused HA1 recombinant protein was purified through the combined use of anion-exchange and glutathione affinity chromatography. The purified protein was assessed by SDS- PAGE and western blot analysis. As shown in Fig. 1A, the recombinant HA1 protein fused to GST indicated a single band with a molecular weight of 62.5 kDa in the SDS-PAGE and western blot analyses, within which the predicted size of HA1 was about 36.5 kDa. Activity of the recombinant HA1–GST fusion protein was measured by hemagglutination assay, which is based on an activity of viral HA that binds to and aggregates avian and mammalian erythrocytes (KILLIAN, 2008). This hemagglutination assay is a convenient and rapid method for determining the level of influenza virus present in a sample (Rimmelzwaan et al., 1998). To investigate hemagglutination activity, agglutination of chicken RBCs was carried out by mixing 1% chicken RBCs (v/v) and serially diluted HA1 protein (10 μg to 0.005 μg) at room temperature. Figure 1B provides a top-down view of the hemagglutination reaction mixture in a round-bottomed 96-well plate. BSA did not agglutinate the erythrocytes to the same degree as HA1. In contrast, the recombinant HA1 purified from E. coli efficiently agglutinated the erythrocytes, even though it was still fused with GST. At a low HA1 concentration, RBCs were observed at the bottom of the well. However, at a high concentration of HA1, the formation of RBC–HA complexes prevented the sinking of RBCs. The lack of RBCs at the bottom of the well provides substantial evidence of hemagglutination activity by the purified HA1–GST fusion protein. Therefore, we confirmed the biological activity of our purified HA proteins, and that the proteins were legitimate for use in the subsequent aptamer screening process (SELEX).

Purification of biologically active HA1.
Selection of RNA aptamers specific to hemagglutinin HA1 subunit
To isolate high-affinity RNA aptamers against the HA1 subunit, we first designed a random DNA template to produce an RNA library. RNA library oligonucleotides containing the 40 random sequences of nucleotides were prepared by PCR and in vitro transcription (Fig. 2A). Fifteen iterative rounds of selection were conducted, increasing the stringencies of RNA binding to the target protein as the rounds progressed. Starting from the 5th round, more stringent conditions were employed by reducing the protein concentration at each subsequent round: 25 μg (rounds 5–6), 10 μg (rounds 7–8), 5.0 μg (rounds 9–10), 2.5 μg (round 11–12), and 1.25 μg (rounds 13–15). After fifteen cycles of selection, the RNA pool of the 15th round was cloned and 15 individual aptamer clones were sequenced. Sequence alignment of these clones enabled grouping of the clones into three groups based on sequence similarity (Fig. 2B). Each group had a characteristic sequence that might be considered as a binding site to the HA1 protein; there were conserved sequences of five nucleotides (AGAAG), and eleven nucleotides (AAGGG) followed by (AGUUGA) in groups 1 and 2, respectively. The five clones in group 3 were found to contain a different conserved sequence of five nucleotides (AAUCA). The secondary structures of eight different HA1 aptamer candidates were predicted using the RNA secondary structure prediction program, Mfold, which is based on the Zuker algorithm (ZUKER, 1989). All of RNA aptamers were shown to contain a number of stem-and-loop structures, in which the conserved sequences mainly reside at the stem-and-loop region in the RNA secondary structure (Fig. 3).
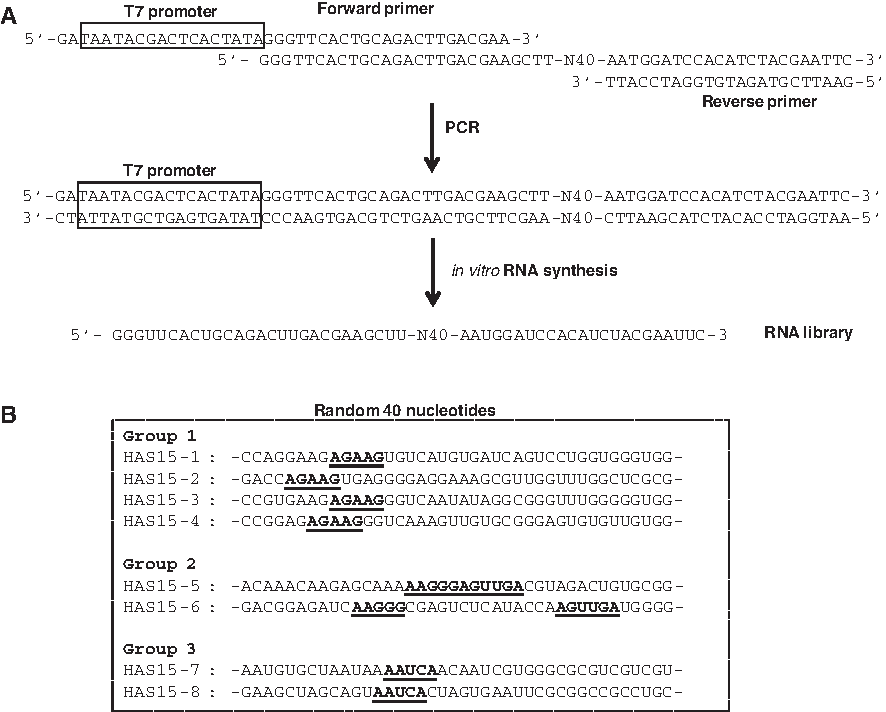
Preparation of oligonucleotides for in vitro selection and sequences of selected RNAs.

Secondary structures of the HA1 RNA aptamer candidates. The predicted secondary structures of the eight different sequences of the HA1 aptamer candidates (HAS15) were obtained using the Mfold program based on the Zuker algorithm. The conserved sequences in each group are indicated by shading on each structure.
After 15 rounds of the selection process, HA1 binding affinity of the 15th-round RNA pool was compared with that of the RNA pools from the 0th and the 7th rounds through the semiquantitative RT-PCR amplification of the RNAs bound to the target protein (Fig. 4A). The RNA pools of each round were loaded onto a chromatography column charged with the HA1 protein. Both the flowthrough fractions (HA1-unbound RNAs) and affinity-eluted fractions (HA1-bound RNAs) were collected and subjected to RT-PCR. PCR was limited to 13 cycles to avoid saturation of cDNA amplification. The cDNAs were visualized by ethidium bromide staining under ultraviolet (UV) illumination on an agarose gel, which reflected the amount of RNA that had bound to HA1. The RNA from the 15th-round pool was bound to HA1 more prominently than that from the 0th- and 7th-round pools as well as the negative control containing irrelevant RNA sequences (Fig. 4B). Therefore, RNAs obtained at the 15th round of SELEX exhibited a significant affinity for the HA1 subunit of HA.
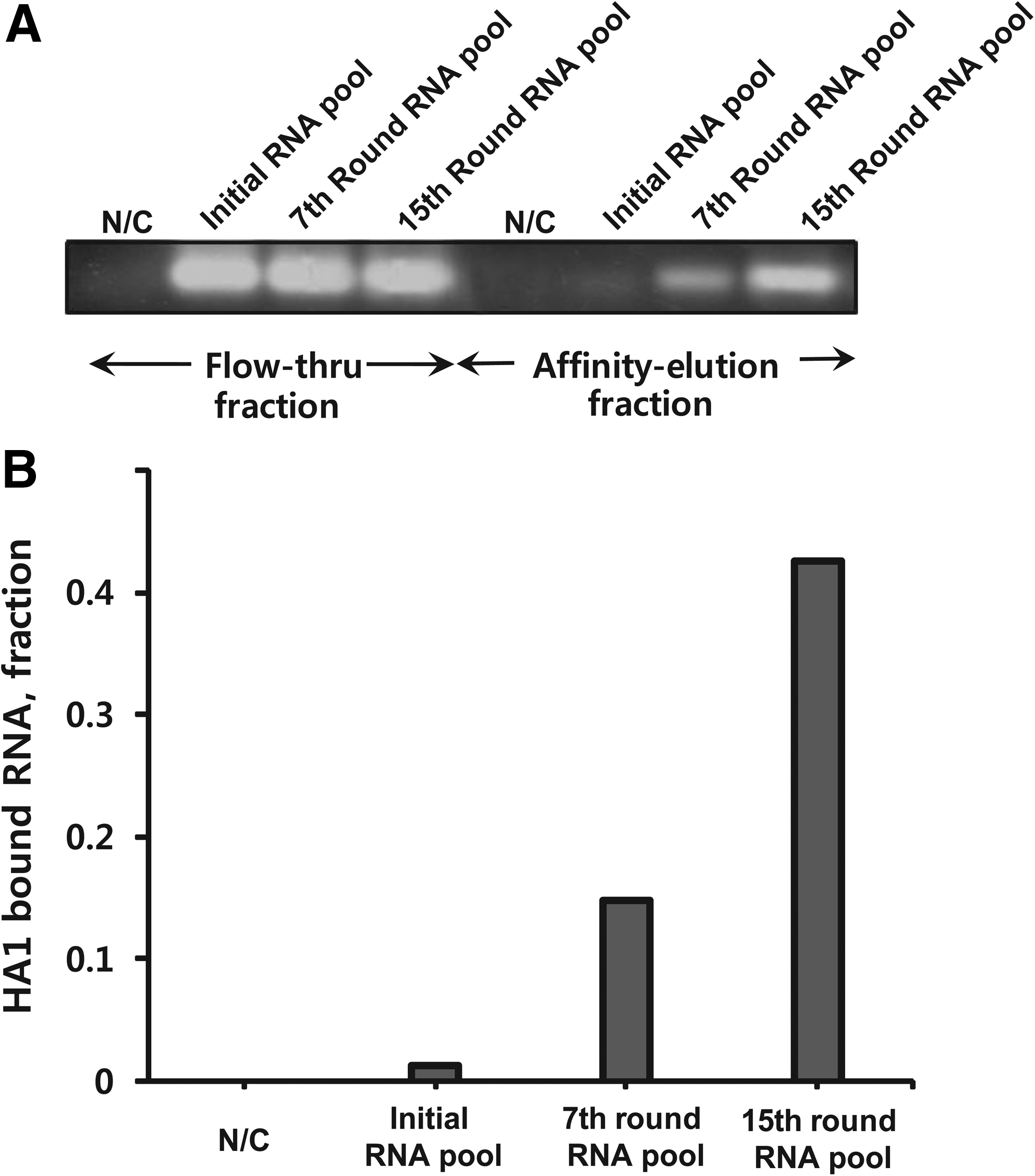
Measurement of the RNA bound to HA1.
Evaluation of antiviral activity of the selected aptamer
To evaluate the inhibitory activity of the RNA aptamer candidates against viral HA1 protein, we performed a hemagglutination inhibition assay. First, the eight different RNA sequences selected as aptamer candidates (Fig. 3) were synthesized and compared in the assay, to select the most effective aptamer. HA1 protein (1.2 μg) was serially diluted by a factor of 4/5, and 1 μg of each RNA aptamer candidate was added to each separate well. As shown in Fig. 5A, most of the aptamer candidates did not exhibit a strong inhibitory effect against HA1 induced erythrocyte agglutination, except for one RNA candidate, HAS15-5. HAS15-5 displayed a significant degree of inhibition of HA1 hemagglutination, compared with the other RNA aptamer candidates, which suggests that HAS15-5 effectively binds and inhibits the viral HA1 protein and prevents erythrocyte agglutination.

Hemagglutination inhibition assay for the evaluation of the antiviral activity of the selected RNA aptamer.
Next, to confirm the antiviral effect of the HAS15-5 aptamer against avian influenza virus, we inoculated the virus into SPF eggs with or without HAS15-5. For this method, we used 9- to 11-day-old SPF eggs as virus incubators. During incubation, the virus replicates in the cells that make up the chorioallantoic membrane. New virus particles are produced by budding and are released into the allantoic fluid. Allantoic fluid harboring the virus was harvested and mixed with designated RNAs at room temperature for 30 minutes. The virus–RNA mixture was injected into the SPF eggs and incubated for 3 days at 37°C. The allantoic fluid was then withdrawn and the hemagglutination activity of the virus fluid was investigated by RBC agglutination assay. As shown in Fig. 5B, the allantoic fluid containing the virus and HAS15-5 exhibited highly suppressed levels of RBC agglutination compared with the allantoic fluids including either the initial RNA pool or the 7th-round RNA pool. This result further suggests that the HAS15-5 RNA aptamer effectively binds to and inhibits the viral HA protein, which results in the suppression of viral replication by RNA aptamer HAS15-5.
Discussion
In this study, we purified a recombinant HA1 protein expressed in E. coli, for the selection of effective RNA aptamers. The purified HA1 protein was shown to exhibit erythrocyte agglutination activity, which is crucial for the use of the protein in the aptamer selection process and for any subsequent aptamer activity assays. To select inhibitory RNA aptamers specific to HA of the avian influenza virus, it is crucial to purify a biologically active HA protein. The HA1 subunit of HA has previously been expressed in and purified from E. coli strain BL21(DE3) for a different DNA aptamer selection study (Cheng et al., 2008). However, the expressed recombinant protein was an insoluble inclusion body, which requires denaturation followed by a compulsory refolding process with 6 M guanidine. Because the refolded HA1 protein was not tested for hemagglutination activity in vitro, the DNA aptamer for the HA1 protein selected in the previous study was not guaranteed to exhibit antiviral activity via the inhibition of HA activity. To overcome any doubts regarding the authenticity of the recombinant HA1 protein arising from the refolding process, we conducted purification of the HA1 subunit using the GST fusion system, which promotes solubility of expressed recombinant proteins.
After 15 rounds of iterative SELEX cycles, we found that the selected aptamer candidates contain short conserved sequences in the loops or stem structures flanking loops in RNAs (Fig. 3). We believe that the conserved sequences in each RNA aptamer candidate are exposed, which facilitates interactions with the HA1 protein. It has been shown that selected RNA aptamers for target proteins often contain a conserved sequence of several nucleotides in the loop structure (Fukuda et al., 2000; Jang et al., 2008). Thus, the conserved sequences residing at the RNA aptamer loop and stem could constitute a binding motif structure to the HA1 subunit of HA, whereas the stem structure of any other sequence could have merely a stabilizing function in the RNA aptamer secondary structure.
HA1 binding assay was performed with the RNA pools from several rounds of SELEX process (Fig. 4). Although the RNA pools of the 15th round exhibited strong binding to HA1 protein, most of aptamer candidates from the 15th-round pool were not able to inhibit hemagglutination of erythrocytes except the HAS15-5 aptamer (Fig. 5). These results suggest that binding of the RNA aptamer to HA is not enough to guarantee suppression of hemagglutination activity. The HAS15-5 aptamer among the eight RNA aptamer candidates is the only one that can inhibit the activity of HA. We speculate that the HAS15-5 aptamer strongly binds to the critical site of HA protein so that the RNA aptamer readily inhibits hemagglutination activity. Further analysis is needed to elucidate the exact binding site(s) of the HAS15-5 aptamer to the HA protein. Alternative to this explanation, a significant degree of inhibition of HA1 by the HAS15-5 aptamer compared to other individual aptamers may result from the different HA1-binding affinities of each aptamer candidate. This hemagglutination inhibition assay was performed by incubating each individual aptamer clone from the 15th-round RNA pool with the protein, in which the HAS15-5 aptamer may exhibit the strongest binding affinity to HA protein.
Previously, DNA aptamers targeting HA of the H5N1 influenza virus were shown to have antiviral activity by enhancing host-cell viability when the host cells pretreated with DNA aptamer were challenged with the influenza virus (Cheng et al., 2008). Despite antiviral activity of suppressing virus replication in host cells by DNA aptamers, direct evidence of inhibition of HA activity by the DNA aptamer was lacking. Thus, in our study the efficacy of HAS15-5 was further evaluated by a hemagglutination inhibition assay using the allantoic fluid of SPF eggs inoculated with type H5 influenza virus and the RNA aptamer. The selected RNA aptamer HAS15-5 significantly inhibited in vitro HA activity by blocking the receptor-binding domain of HA.
In conclusion, the RNA aptamer that was generated in our study, which specifically targets the receptor-binding domain of HA, has antiviral activity by presumably disrupting virus entry to the host cells. There have been no vaccines to prevent the emergence of new viral infections, thus an RNA aptamer could be an attractive therapeutic agent that is comparable to antibodies (Nimjee et al., 2005). Further studies are underway to combine the selected RNA aptamer with an appropriate RNA delivery vehicle, which is mandatory for therapeutic use of RNAs in animal models.
Footnotes
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea Government (MEST) (2010-0019306, 2011-0016385) and a grant from the BioGreen 21 Program (20080401034026), Rural Development Administration, Republic of Korea.
Author Disclosure Statement
No competing financial interests exist.