
Abstract
MicroRNAs are endogenous small non-coding RNAs that regulate gene expression by interfering with translation or stability of target transcripts. The importance of microRNAs for maintaining biological functions is illustrated by the fact that microRNAs are exploited in nature to regulate phenotypes, and by the diverse disease phenotypes that result when microRNAs are mutated or improperly expressed. Disease-associated microRNAs might therefore represent a new class of therapeutic targets. With the recent demonstration that inhibition of miR-122 reduces viral load in hepatitis C patients, microRNA modulators are no longer merely theoretical, but rather, have become strong candidate therapeutics. The complexity of microRNA biology offers a novel mechanism of action for therapeutic intervention but also poses unique challenges for the development of therapeutic modulators as drugs.
Introduction
Epstein-Barr virus (EBV) is a gamma-herpesvirus that establishes lifelong latent infection in >90% of the human adult population worldwide. Latent EBV infection is linked to the development of multiple cancers, and the resulting cancers display distinct patterns of expression of viral proteins and microRNAs. A set of 3 microRNAs derived from the primary EBNA2 transcript (EBV nuclear antigen 2) contribute to B cell transformation (Seto et al., 2010; Feederle et al.,2011). In addition, a set of viral transcripts, the BamHI A rightward transcripts (BARTs), includes alternatively spliced transcripts that are the templates for as many as 44 microRNAs (Pfeffer et al., 2004; Cai et al., 2006; Zhu et al., 2009). The function of the BART microRNAs in the EBV life cycle and the induction of malignancy is currently unknown, but a number of apoptosis and tumor suppressor genes are potential targets for the BART microRNAs. Recently, EBV infection of epithelial cells in vitro led to anchorage-independent growth, and this altered growth property was associated with down-regulation of transcripts enriched for BART microRNA targets (Marquitz et al., 2012). These transcripts represented molecular functions including cellular movement, cell signaling, cell growth and proliferation, and apoptosis, characteristic of transformation. Therefore, the BART microRNAs are likely contributors to the development of epithelial malignancies linked to EBV. Interestingly, the majority (90%) of the cellular transcripts targeted by the most abundant EBV microRNAs are also targeted by human microRNAs through distinct binding sites in the 3′ UTR (Riley et al., 2012). Many of these are targets of the oncogenic miR-17∼92 cluster, which regulates apoptosis, Wnt signaling, and cell cycle. Host and viral microRNAs therefore co-target oncogenic and apoptotic genes and can have a cumulative effect on targets during viral latency. In addition to encoding its own microRNAs, EBV infection alters the expression of several cellular microRNAs, (Jiang et al., 2006; Cameron et al., 2008a; Cameron et al., 2008b; Godshalk et al., 2008; Lu et al., 2008; Yin et al., 2008; Rosato et al., 2012) including miR-155 and miR-21 to promote growth and predispose cells to transformation, and miR-146a, which functions in the innate anti-viral response.
The use of microRNAs by the KSHV and EBV viruses is an example of how these key regulators are exploitable to alter cellular function. In these cases, microRNAs are exploited to produce disease, but this process is emblematic of how modulating the activity of these molecular regulators can affect broad biologic functions. Learning from nature and exploiting microRNAs as key regulators is an attractive new way to approach dysregulation of cellular function, not through monogenic approaches but through pathways that have evolved to be post-transcriptionally regulated by microRNAs.
Here we review some of the unique complexities of microRNAs, how these complexities impact drug development, and how they are being approached to advance microRNA modulators as therapeutics. In particular, we will consider (1) identifying key functional and pharmacodynamic targets for microRNAs from among the many regulated targets, (2) the influence of biological context on microRNA function and how this impacts microRNA drug target selection and pharmacokinetic/pharmacodynamic (PK/PD) relationships, (3) how the potential for microRNAs to have different targets and functions in different species impacts selection of pre-clinical models, and (4) pharmacological considerations for microRNA modulators.
The microRNAs
microRNAs were first identified in Caenorhabditis elegans as small RNAs that control key developmental transitions (Reinhart et al., 2000; Lee et al., 2001). These regulatory RNAs are endogenous ∼22 nucleotide non-coding RNAs that regulate expression of target genes through sequence-specific hybridization to target messenger RNAs. MicroRNAs have now been identified in plants, algae, and animals (Molnar et al., 2007; Kozomara and Griffiths-Jones, 2011), but have not been identified in yeast. Many microRNAs are conserved across multiple species, indicating their evolutionary importance as modulators of biological functions. A key attribute of microRNAs is that they may regulate gene networks or pathways to control biological functions, playing important roles in differentiation, development, and physiology. MicroRNAs regulate the expression of target transcripts via mRNA degradation or translational arrest (BARTEL, 2009; Carthew et al., 2009; Guo et al., 2010; Huntzinger and Izaurralde, 2011). MicroRNAs bind multiple target mRNAs with partial complementarity, mostly involving the 6- to 8-nt seed sequence within the first 8 nt of the 5′ end of the microRNA guide strand (LAI, 2002; BARTEL, 2009). Seed pairing has been shown to be both necessary and sufficient for target regulation by microRNAs in some experimental contexts (Lewis et al., 2003; Doench and Sharp, 2004; Krek et al., 2005; Lewis et al., 2005), but not in others (Ha et al., 1996; Vella et al., 2004; Didiano and Hobert, 2006; Tay et al., 2008). Recently, alternative modes of target pairing by microRNAs have been described, including centered sites (Shin et al., 2010) and bulged sites (Chi et al., 2012). Since mammalian microRNAs do not require perfect complementarity for target recognition, a single microRNA is able to regulate multiple, perhaps hundreds of, messenger RNAs (Lim et al., 2005; Baek et al., 2008; Selbach et al., 2008). It is estimated that microRNAs as a class regulate the expression of 60% of genes in the genome (Friedman et al., 2009). In therapeutic applications, the specific inhibition of a microRNA or the addition of a single microRNA mimetic may produce a phenotype that is derived from a complex set of gene expression changes.
The targets
MicroRNAs could impact a given phenotype through regulation of a single key target or through concomitant regulation of a subset of targets. In some instances a phenotype can be explained by partial suppression of a single target, as illustrated by the ability of miR-150 to control lymphocyte development by regulating the expression of the seed-matched target c-Myb (Xiao et al., 2007). For other microRNAs the story is more complex, with the phenotype being controlled by the coordinated suppression of multiple targets (Linsley et al., 2007; Georges et al., 2008; Valastyan et al., 2009). MicroRNAs regulate each individual mRNA only modestly (∼30%–50% down-regulation), yet this modest degree of target silencing, is sufficient to induce phenotypic changes.
Because a single microRNA can regulate hundreds of targets, the biological functions of microRNAs are not easily discerned from an examination of their targets. Predictive computer algorithms can assist in the identification of biological targets, but these algorithms predict only ∼50% of the regulated targets detected by global mRNA measurement techniques (Baek et al., 2008). In addition, since different prediction algorithms incorporate distinct aspects of microRNA-target interactions (conservation, accessibility, sequence context, etc.), these algorithms can return disparate sets of predicted targets. The effects of potential microRNA therapeutics are best measured using global mRNA expression methods, such as microarray or RNA sequencing, coupled with statistical techniques that can measure small changes in many genes to identify the significantly regulated, seed-matched targets. This complexity is a challenge in drug development because it is not always clear which (or which combination) of the potential mRNA target(s) are being modulated to produce the phenotypic change of interest (Fig. 1).
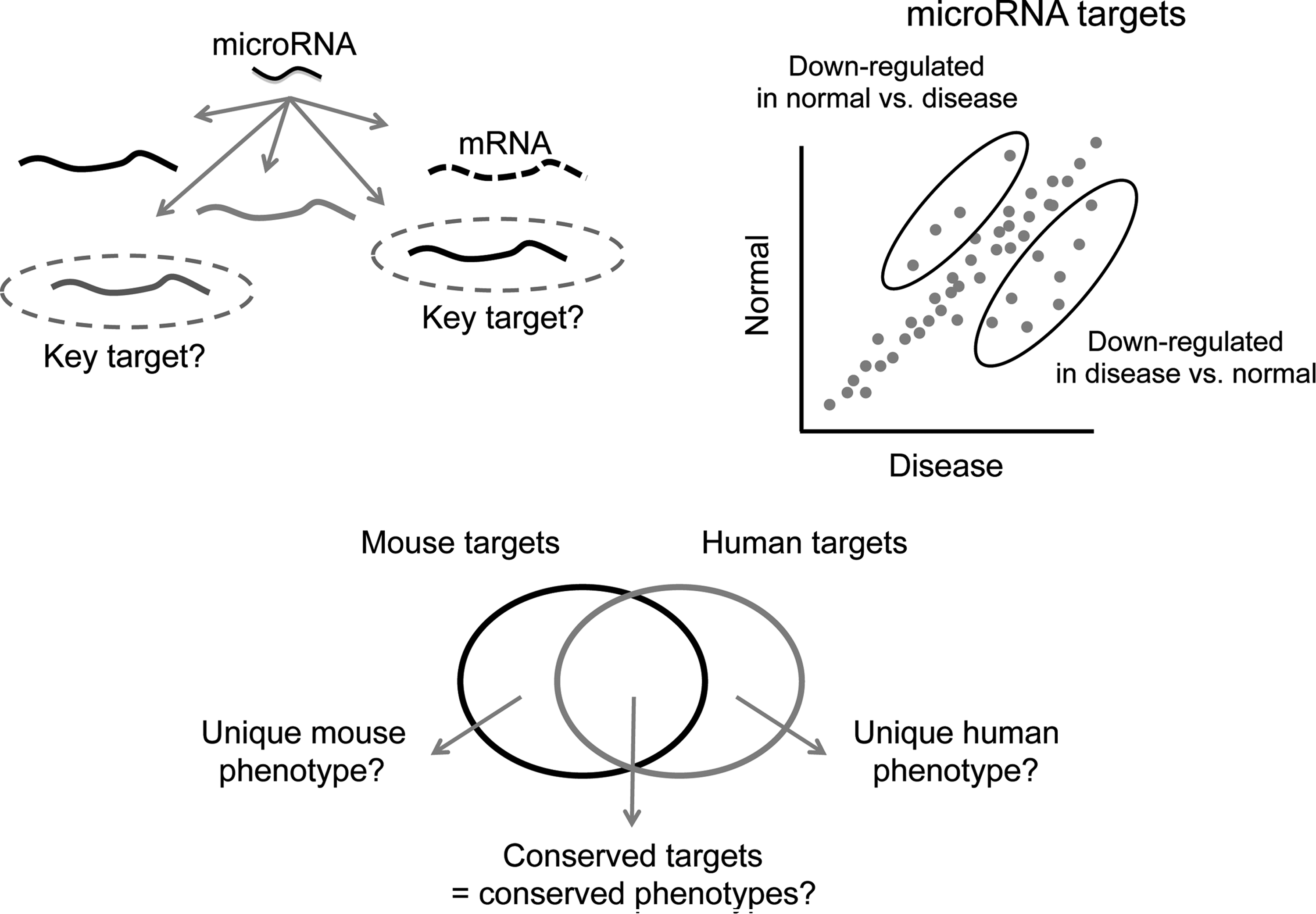
Unique complexities of microRNA biology can impact the selection and development of microRNA therapeutics. The complexity of microRNA biology offers a novel mechanism of action for therapeutic intervention but also poses unique challenges for the selection of microRNA modulators as drugs, including identification of the key pharmacodynamic targets/pathways/networks from among the many transcripts regulated by a microRNA (upper left), the impact of biological context and disease state on target regulation (upper right), and species-specific regulation of targets that can potentially lead to species-specific phenotypes (lower panel), thereby complicating extrapolation from pre-clinical models to man.
The regulation of coordinated gene networks differentiates microRNA modulation as a therapeutic modality and provides a therapeutic advantage reminiscent of combination drug therapy, but in this case the combination has been derived through evolutionary processes. Among the mRNAs regulated in response to microRNA modulation will be the key drivers for the phenotype of interest, in addition to numerous passengers—targets whose altered expression correlates with microRNA modulation but whose regulation may not impact the phenotype. This unique aspect of microRNA biology creates challenges in defining mechanism of action for a microRNA, and in selection of relevant PD markers for therapeutic modulation of microRNA function. Elucidating the driver targets is necessary for optimization around the most relevant activity of the microRNA modulator to facilitate selection of lead sequences, for understanding PK/PD relationships, and for drug development in general.
One approach to identifying driver targets is functional annotation of the direct target transcripts. Some microRNAs target sets of transcripts that function in the same or related pathways, which can provide insight into the biological roles of these microRNAs (Stark et al., 2003; Grun et al., 2005; Lewis et al., 2005; Linsley et al., 2007). Functional analysis of targets using statistical enrichment for biological function (e.g., GO annotation) and network analysis can reveal molecular interactions and biological networks that are impacted by the target transcripts. However, the repertoires of transcripts targeted by some microRNAs are not statistically enriched for specific biological functions or processes, or enrichment might involve only a minority of the targets. It can therefore be difficult to determine solely from functional annotation which target(s) are responsible for microRNA phenotypes and this leaves experimental approaches as the best way to address this question.
Further complicating studies of microRNA mechanism of action, a given microRNA might regulate different targets and display diverse functions in different cellular contexts depending on the repertoire of direct targets expressed in that context (e.g., disease state, Fig. 1)(Sood et al., 2006). For this reason, microRNA targets and PD biomarkers are best defined experimentally in the relevant biological context. This fact may drive more aspects of the drug discovery process into relevant disease models because PK/PD relationships for target de-repression (or repression) may not be the same in the normal and the disease states.
Approaches to identify phenotypic drivers by target disruption, such as small interfering RNA (siRNA-mediated) gene repression or mouse knockouts, can demonstrate that gene loss of function plays a causal role in the disease phenotype. Since microRNAs typically mediate transcript repression in the range of 30%–50%, the 70%–100% repression resulting from siRNAs or knockout mice is not representative of a microRNA-mediated phenotype. Only if a change in expression level equivalent to microRNA-mediated silencing phenocopies the microRNA can the transcript be declared the driver target of the microRNA. Functionally defining the driver target(s) for a microRNA-mediated phenotype is best accomplished by titrating the target transcript repression to the same magnitude that is achieved by the microRNA. Examples of this approach include use of heterozygous animals for 50% loss of function (Xiao et al., 2007), and titrating siRNA concentration for low-magnitude transcript silencing (Linsley et al., 2007). The same concept applies to overexpression studies to phenocopy target de-repression by an anti-miR. To date, there are limited reports of microRNA mechanism of action in disease settings. Despite this, differences in approaches to defining key targets, combined with the potential for regulation of different mechanisms in different biological contexts, has resulted in diverse definitions of function for any particular microRNA. A few examples are presented to illustrate this point.
miR-10b
miR-10b promotes invasion and metastasis by targeting HOXD1 (Ma et al., 2007; Bourguignon et al., 2010) or TIAM1 (Moriarty et al., 2010) in breast cancer, HOXD1 in gastric cancer (Liu et al. 2012b), neurofibromin in neurofibromatosis type 1 (Chai et al., 2010), KLF4 in esophageal cancer (Tian et al., 2010), and HOXD1 (Sun et al., 2011) or CDKN1A, CDKN2A, BCL2L1, and TFAPC (Gabriely et al., 2011) in glioblastoma. The growth-promoting role of miR-10b in glioblastoma has been further dissected to suggest that repression of different targets triggers cell survival and proliferation in different glioblastoma cells depending on their genetic and epigenetic contexts (Gabriely et al., 2011). Contrasting the role of miR-10b as an oncomiR, one study suggests a tumor suppressive role for miR-10b in gastric cancer, where epigenetic silencing of miR-10b results in increased expression of oncogenic MAPRE1, confirmed by decreased colony formation in gastric cancer cells upon transfection with miR-10b precursor (Kim et al., 2011).
miR-375
miR-375 was originally identified as a pancreas-specific microRNA, where it functions in maintaining alpha and beta cell mass and regulates insulin secretion (Poy et al., 2004; Kloosterman et al., 2007; Avnit-Sagi et al., 2009; Poy et al., 2009). Subsequently, miR-375 was shown to be expressed in multiple tissue contexts, and its dysregulation is associated with multiple disease states. miR-375 loss is associated with development of cancers and exerts its tumor suppressive effect through regulation of AEG-1 (He et al., 2011) or YAP in liver cancer (Liu et al., 2010), AEG-1 in head and neck squamous cell carcinoma (Nohata et al., 2011), YAP1 in lung cancer (Nishikawa et al., 2011), lactate dehydrogenase B in maxillary sinus squamous cell carcinoma (Kinoshita et al., 2012), SP1 in cervical cancer (Wang et al., 2011), IGF1R (Kong et al., 2012) or PDK1(Li et al., 2011) in esophageal cancer, and JAK2 (Ding et al. 2010) or PDK1 and 14-3-3ζ (Tsukamoto et al., 2010) in gastric cancer. Interestingly, in contrast to a tumor suppressive role in other cancer settings, miR-375 is reported to be oncogenic in breast cancer, by regulating a positive feedback loop with estrogen receptor alpha (de Souza Rocha Simonini et al., 2010). In addition, miR-375 is reported to be up-regulated in prostate carcinoma, where it targets SEC23A to promote cell proliferation (Szczyrba et al., 2011).
let-7
Let-7 (lethal-7) is a family of 8 microRNAs (let-7a–let7i) that share identical seed regions but have divergent sequences in the 3′ portion of the mature microRNA. Members of the let-7 family are tumor suppressive in prostate cancer via regulation of Myc and downstream androgen receptor (Nadiminty et al., 2012) or by targeting E2F2 and CCND2 (Dong et al., 2010), by targeting HMGA2 and RAS in lung cancer (Kumar et al., 2008), RAS in pancreatic cancer (Ali et al., 2012), estrogen receptor α36 in breast cancer (Zhao et al., 2011), Bcl-xL in hepatocellular carcinoma (Shimizu et al., 2010), basigin in melanoma (Fu et al. 2011), or MYCN in neuroblastoma (Buechner et al., 2011). In contrast, let-7 family members promote cancer development by targeting MYH9 in gastric cancer (Liang et al., 2011) or FAS in colon cancer cells (Geng et al., 2011). But the function of let-7 is not restricted to controlling cancer development. Let-7 also plays a central role in control of mammalian glucose metabolism. Global or tissue-specific overexpression of let-7 in mice leads to insulin resistance and impaired glucose tolerance through regulation of multiple components of the insulin-PI3K-mTOR pathway (Frost and Olson, 2011; Zhu et al., 2011), suggesting antagonism of let-7 as a potential treatment for type 2 diabetes.
That microRNAs have different, and sometimes opposing, functions in different cellular/tissue contexts is an important consideration in selecting the microRNA to target therapeutically. Another factor to consider when selecting the microRNA drug target is the expression of the microRNA itself. Many microRNAs exist within families of microRNAs that share a seed region but differ in the microRNA sequence outside of the seed region. The relative expression levels of family members might differ in different cell types, in different tissues, and even across species. A particular microRNA modulator might therefore have different phenotypic consequences in different disease states, as well as between pre-clinical models and humans. Our knowledge of microRNA expression and function in various disease settings and different species is increasing rapidly, enabling selection of drug targets with higher probability of success.
Species-specific effects of microRNAs
There is another unique feature of microRNAs that must be taken into consideration for selection of the microRNA drug target and for selection of pre-clinical models for drug development: the potential for species-specific effects (Fig. 1). Although microRNAs tend to be highly conserved across species, there might be less conservation of the transcripts targeted by microRNAs. While the majority of functionally characterized binding sites for microRNAs reside in 3′ UTRs (untranslated regions), microRNAs can also regulate target transcripts through binding to 5′ UTRs or coding regions of target transcripts (Nakamoto et al., 2005; Miranda et al., 2006; Easow et al., 2007; Duursma et al., 2008; Forman et al., 2008; Tay et al., 2008; Chi et al., 2009; RIGOUTSOS, 2009; Zhou et al., 2009; Hafner et al., 2010). A recent large-scale data analysis identified abundant conserved microRNA target sites in the 5′ UTR and coding sequence (Zhou et al., 2009), suggesting that microRNA-mediated regulation is not limited to the 3′ UTR. MicroRNA targets sites in the coding region can trigger target regulation, although less effectively than target sites in the 3′ UTR (Easow et al., 2007; Hafner et al., 2010). These data suggest that 3′ UTRs of target transcripts may be more critical for functional targeting, but other binding sites may play more subtle regulatory roles. It has been hypothesized that the 3′ UTR sequences are under selective pressure to maintain microRNA binding sites such that these sites should be more conserved than random sequences (Chen and Rajewsky, 2006b). Indeed, microRNAs and some 3′ UTR motifs are conserved, but 3′ UTR sequences and some microRNA-target relationships are divergent across species (Chen and Rajewsky, 2006a). Therefore the transcripts targeted by a microRNA, and thus the phenotypic consequences of microRNA modulation, have the potential be different in different species. These differences have obvious consequences for the selection of appropriate pre-clinical models for drug development and require some understanding of the relationship between target mRNAs in laboratory animals and those in humans.
The let-7 gene was initially discovered as an essential developmental gene in C. elegans. It was later identified as one of the first microRNAs (Reinhart et al., 2000; Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001), and is now known to be highly conserved in both sequence and developmental expression in organisms spanning wide evolutionary distance (Pasquinelli et al., 2000; Lagos-Quintana et al., 2003; Pasquinelli et al., 2003; Lee et al., 2007). Biological functions for let-7 in animals are diverse and include the regulation of stem-cell differentiation, neuromusculature development, limb development, and cell proliferation and differentiation (reviewed in Roush and Slack, 2008). Moreover, many let-7-family members function as tumor suppressors in a variety of cancers (Esquela-Kerscher and Slack, 2006; Shi et al., 2008). An intriguing feature of the conservation of let-7 function is the presence of a core set of conserved targets, while the overall target repertoire of let-7 is strikingly divergent across evolution, developmental timing, and cancer (Nimmo and Slack, 2009).
The non-conserved targets of the highly conserved let-7 microRNA family are an example of divergence and repurposing of a microRNA by different species. In C. elegans, let-7 controls developmental transitions mainly in hypodermal cells (Reinhart et al., 2000). In contrast, the main function of let-7 in Drosophila seems to be to control events in the development of the adult nervous system (Sokol et al., 2008). In particular, let-7 is required for remodeling of the fly neuromusculature during the transition from larva to adult. A key let-7 target in this event in the fly is Abrupt (Sokol et al., 2008), which is not a target of let-7 in worms or mammals. In mammals, a key target of let-7 is the oncogene HMGA2 (Lee and Dutta, 2007; Mayr et al., 2007). The orthologs of HMGA2 are not targets of let-7 in worms or flies. Therefore, while let-7 functions in controlling differentiation and cell fate in these diverse species, the specific developmental pathways that are regulated, and the set of key direct targets of let-7, differ in the worm and the fly (Grun et al., 2005; Lall et al., 2006) and in humans (Lee et al., 2007; Mayr et al., 2007).
A recent study by Liu et al. (Liu et al., 2012a) approached the question of species-specific target regulation by comparing 3′ UTR sequences and microRNA-mediated regulation of a particular target transcript in different species. The study reports that the 3′ UTR sequences of the progesterone receptor (PGR), and associated microRNA binding sites, are conserved among primates, but poorly conserved between primates and rodents. The 3′ UTR of the rhesus monkey PGR contains 33 potential binding sites for 15 microRNAs. Among these sites, 7 were rhesus monkey specific and 26 were conserved only in primates. PGR was confirmed to be a direct target of the highly conserved miR-96 in humans and rhesus monkeys, but not in rodents. Furthermore, PGR could be regulated by miR-375 in rhesus monkey but not in humans or rodents. The same study demonstrates that miR-219-5p indirectly regulates PGR expression through a primate-specific long-noncoding RNA (lncRNA), suggesting an additional primate-specific mode of PGR regulation by a microRNA. Some lncRNAs are reported to harbor microRNA binding sites (Poliseno et al., 2010; Braconi et al., 2011), making them direct targets of microRNA regulation in some cases. Most lncRNAs are subject to a low degree of sequence constraint and are less conserved than protein coding genes (Pang et al., 2006; Marques and Ponting, 2009). MicroRNA-mediated regulation of lncRNAs could represent another mechanism for species-specific target recognition. A unifying hypothesis of competing endogenous RNA activity has been proposed to explain how mRNAs, transcribed pseudogenes, and lncRNAs communicate with each other via microRNA response elements to form a large-scale regulatory network across the transcriptome (Salmena et al., 2011). The hypothesis asserts that coding and non-coding RNAs can compete for microRNA binding, thereby communicating to each other to regulate their respective expression levels. Coding and non-coding RNAs that share multiple microRNA binding sites would be expected to crosstalk, or compete, effectively. The ability of multiple types of RNAs to compete for microRNAs, in addition to species-specific expression of coding and non-coding RNAs, would clearly add complexity to the use of microRNAs as therapeutic targets.
Another example of species-specific target regulation by microRNAs is provided by the miR-430/427/302 microRNA family expressed during early embryogenesis. These microRNAs share an identical seed region as well as a high degree of sequence similarity in the 3′ region of the microRNA, and thus can be considered as evolutionarily conserved members of the same microRNA family. Despite the evolutionary conservation of these microRNAs, they display different embryological activities in human, Xenopus laevis, and zebrafish as a result of species-specific target selection and availability (Rosa et al., 2009).
Species-specific target regulation suggests that there could be differences in phenotypic responses to microRNA modulators between animal models and man, complicating the extrapolation of pharmacologic effects from laboratory animals to man. One possible approach to overcoming this hurdle would be to demonstrate that target transcripts, particularly driver targets, for the phenotype of interest are regulated consistently across species and/or to confirm that the molecular responses of interest occur in human cells or tissue preparations. Another provocative idea is that the phenotypic consequence of a microRNA modulator could be the same in different species even if the individual target transcripts contributing to that phenotype are overlapping but not identical in different species. The development of microRNA modulators as therapeutics would be facilitated by demonstrating cross-species efficacy as early as possible in the drug development process.
In the best characterized example to date, inhibition of miR-122 in species from mouse to man has produced phenotypic changes that are remarkably similar, with reductions in cholesterol as the major phenotypic change. Even with the overexposures that define toxicity studies (Hildebrandt-Eriksen et al., 2012), the phenotypic changes observed in primates did not differ from the changes seen at pharmacologic doses in mouse (Esau et al., 2006; Elmen et al., 2008), monkey (Elmen et al., 2008), chimpanzee (Lanford et al., 2010) and man (Janssen et al., 2011). If and when the appropriate human liver biopsies become available, it would be instructive to compare the patterns of gene expression changes at the transcriptional levels across the various species with those of man. Clearly, as more microRNA-targeting drugs enter large-scale non-clinical and clinical trials, it will be useful to continue to compile the cross-species comparisons in vivo and to establish how well the in silico predictions and in vitro models predict in vivo responses.
Table 1 summarizes the complexities of microRNA biology that have been discussed thus far, the impact these complexities have on clinical development of microRNA-based therapeutics, and potential strategies to improve understanding of microRNA function and enhance the probability of success of microRNA drugs.
ASO, antisense oligonucleotide; PK/PD, pharmacokinetic/pharmacodynamic; siRNA, small interfering RNA.
Dose response and microRNA therapeutics
The dose response relationship is one of the key principles of pharmacology dating back to the time of Paracelsus (“Dosis facit venenu”—dose makes the poison). For microRNA-targeting therapeutics, there have been clear demonstrations of dose–response relationships in various animal models with various microRNA targets. Using anti-miRs or antagomiRs to inhibit miR-122, multiple laboratories have demonstrated dose-dependent pharmacologic effects in species ranging from mouse to man (Krutzfeldt et al., 2005; Esau et al., 2006; Elmen et al., 2008; Elmen et al., 2008; Santaris unpublished observation). Dose responsiveness has been described for multiple other targets as well, in both cell culture systems and in vivo (Obad et al., 2011; Hullinger et al., 2012). Thorough characterization of the dose response relationships in vivo may be complicated by the reported non-linear pharmacokinetics of phosphorothioate oligonucleotides (Levin et al., 2008; Geary et al., 2008), as well as by the biology of microRNA/RNA induced silencing complex (RISC)-mediated effects in which the bottom portion of the typical sigmoidal curve may be very shallow owing to the relatively small changes in target de-repression observed even at maximal inhibition. Thus, dose response curves may take on the appearance of an all-or-none type of response simply because of the subtlety of the responses.
At the higher end of the dose response curve, like the more typical pharmacology of small molecule drugs, the pharmacologic effects of microRNA-targeting drugs should be saturable as the receptors become fully occupied. In the case of microRNA inhibitors, activity is dependent on the binding and sequestration of the target microRNA in an inactive microRNA/anti-miR heteroduplex. The receptor in this case is the targeted microRNA. When all of the target microRNA in the cell is sequestered, addition of more anti-miR will not induce additional pharmacology but can induce additional non-specific effects. The concentration of anti-miR that is required to produce the maximal sequestration of the target microRNA is a function of the number of copies of the target microRNA in the cell, the affinity of the anti-miR for the target, and the concentration of the freely available anti-miR in the cell. The latter is a function of the amount of anti-miR available in the cytoplasm, not bound in endosomes or bound by cytoplasmic proteins. Using high affinity anti-miRs would be expected to force the equilibrium between protein binding and target microRNA binding more toward productive microRNA binding. However, the definitive proof of this hypothesis is yet to be made, owing to the complexities of measuring functional microRNA/anti-miR complexes in specific cellular compartments without disrupting the cell. Experiments attempting to monitor functional microRNA/anti-miR interaction by failure to amplify or probe the target microRNA in cell or tissue lysates can be difficult to interpret. Upon lysing the cell and disrupting potential subcellular compartmentalization or protein binding of the anti-miR, artificial microRNA/anti-miR complexes that did not exist in the native state of the cell might be observed. Therefore, these experiments cannot provide conclusive information on dose dependence of functional microRNA inhibition or saturation by an anti-miR. In any case, determining the saturating concentration or the dose of anti-miR required to produce saturation is critical to dose setting in clinical trials where the desire is to produce the pharmacologic effect of interest at the lowest dose possible to avoid non-specific effects.
For microRNA mimetics, in which the mature microRNA is replaced or expressed in a cell, the dose response relationships will be similar to those observed for exogenous siRNAs. Typical dose response relationships for exogenous siRNAs have been reported in cell culture (Vaishnaw et al., 2010) and in vivo in species from mouse to man (Soutschek et al., 2004; Alnylam, unpublished observations). The pharmacology of microRNA mimetics would be expected to be limited by the available RISC. Once these complexes have been completely occupied with the microRNA mimetic, pharmacology would be expected to saturate. The potentially adverse effects of full saturation of RISC and other related complexes were demonstrated by the overexpression of an shRNA in mice (Grimm et al., 2006) with lethal consequences. Whether exogenous microRNA mimetics can be delivered at concentrations that achieve those resulting from shRNA overexpression (Grimm et al., 2006) has yet to be established, but these results suggest the nature of the effects of RISC saturation and other microRNA processing activities that could be expected in the extreme.
An additional word on microRNA mimetics
microRNA mimetics represent an additional level of complexity compared to anti-miRs. On the one hand, there are the RISC saturation effects described by Grimm et al (Grimm et al., 2006). On the other hand, there is the potential to induce unwanted effects when novel microRNAs are introduced into a cell. One approach to avoid the latter problem is to administer mimetics only for microRNAs that are ubiquitously expressed, thereby avoiding unwanted and unanticipated effects in a cell(s) that has never expressed that microRNA, and only replacing activity needed to produce the pharmacology of interest in a target cell type that has lost the function of that microRNA. The field of microRNA mimetic toxicity is generally unexplored at this time. A study by Khan et al. (Khan et al., 2009) might provide some insight into unexpected effects associated with RISC saturation. Khan et al. performed meta-analyses to conclude that siRNAs transfected into cells could compete with endogenous microRNAs for association with RISC, resulting in de-repression of microRNA targets. (Khan et al., 2009). The in vivo demonstration of siRNAs affecting the function of endogenous microRNAs has not yet been described in the literature. Notably, a study by John et al. demonstrated that synthetic siRNAs delivered to the livers of mice and hamsters in vivo produced effective target gene silencing without de-repression of 7 target transcripts for the hepatocyte microRNA miR-122 (John et al., 2007). It remains to be determined whether further experiences with siRNA pharmacology in vivo including humans, as well as direct studies of microRNA mimetics, includes saturation of RISC to affect the functions of endogenous microRNAs. Bioinformatics analyses of the transcriptome of tissues from animals treated with siRNA or microRNA mimetics will ultimately address this question.
Putting it into Practice
While there are complexities associated with the targeting of microRNAs for therapeutic purposes, there are many reasons why it is attractive to explore microRNA modulation as a therapeutic approach. The importance of the broad regulation of expression by non-coding RNAs is now becoming more appreciated and it is clear that non-coding RNAs represent a significant “investment” of the genome in higher species. As such, non-coding RNA is likely a good target for intervention. Assuming that these regulatory microRNAs are worth exploring as therapeutics, on a practical level, what therapeutic modalities are being used to exploit the therapeutic potential of microRNAs?
To date, the therapeutic modalities used for the inhibition of microRNA function (antagonists) or supplementation of microRNA function (agonists) are oligonucleotides: single stranded oligonucleotide complementary to the target microRNA of interest for inhibition (anti-miRs) or duplex oligoribonucleotides corresponding to mature microRNA or microRNA precursor (mimetics). The antagonists and the agonists have very different pharmacologic properties. The single stranded oligonucleotide anti-miRs are generally phosphorothioated and share many of the properties of the single stranded antisense oligonucleotides that have been the subject of multiple reviews (Crooke, 2008). Single stranded oligonucleotides are administered parenterally in the absence of a delivery vehicle. They can be injected or infused and because they are bound by plasma proteins, they are spared from renal filtration and excretion. Uptake by multiple cell types is relatively rapid and plasma is cleared by uptake into multiple organs with liver and kidney having the greatest avidity for uptake. The pharmacokinetics and toxicity of single stranded phosphorothioated oligonucleotides have been reviewed extensively (Geary et al., 2008; Henry et al., 2008; Levin et al., 2008). Unlike the related antisense molecules that generally inhibit translation by directing the cleavage of their cognate mRNA ligands, microRNA inhibitors have been designed to bind to and sequester their targets and do not have to support cleavage enzymes like RNase H. This difference allows microRNA inhibitors to dispense with the oligodeoxynucleotide regions thus allowing the addition of more high affinity modifications to enhance sequestration, even allowing high affinity molecules as short as 7 nucleotides (Obad et al. 2011). Thus, anti-miRs tend to be more highly chemically modified to enhance binding affinities.
Similar to antisense molecules that have been used therapeutically for more than a decade, single stranded microRNA inhibitors can be used to target microRNAs in the liver and kidney without any specific uptake or targeting formulations. Recently, heart and other organs with less efficient oligonucleotide uptake than the liver and kidney have nonetheless been shown to have sufficient uptake to produce phenotypic changes in laboratory animals as a result of microRNA inhibition (Montgomery et al., 2011; Hullinger et al., 2012).
More efficient delivery to target organs will reduce the total dose of oligonucleotide drug required to produce pharmacology in some tissues, but the field continues to search for the optimal targeting molecules and non-toxic formulations. Efficient delivery is a general issue for oligonucleotide drugs, and is not unique to microRNA therapeutics.
The question of how to select microRNA targets suitable for therapeutic modulation is going to be a challenge that is unique to microRNAs. MicroRNAs that are selectively overexpressed in disease states are the most amenable to sequestration (inhibition) approaches. For ubiquitously expressed microRNAs, the challenge will be to exploit the avidity of liver and kidney uptake or to develop targeting formulations to favor delivery to specific organs of interest. Because of the limitations of these approaches, it will be a delicate balance to target widely expressed microRNAs for narrow indications without either local delivery (e.g., injection, inhalation, or topical application) or much more specific targeting formulations than are currently available.
MicroRNA mimetics may behave like siRNA molecules that they resemble. Unformulated siRNA molecules tend to be excreted rapidly from circulation because they are filtered from the plasma due to their failure to bind to plasma proteins. It has been hypothesized that their helical nature allows them to be surrounded by an organized shell of water molecules that prevents them from binding to plasma and cell surface proteins. Extrapolating from siRNA to microRNA mimetics suggests that mimetics will have to be delivered in similar formulations.
There is a report of a microRNA entering the circulation via the diet and being detected in target organs like the liver (Zhang et al., 2012) suggesting that some microRNAs or mimetics can be absorbed and can produce phenotypic changes. In principle, these findings suggest that microRNAs may have properties that were not previously described and are unexpected. However, it is not clear if this particular result is due to the absorption of a naked microRNA or that of a microRNA encapsulated in an exosome—the natural equivalent of a lipid formulation. The field appears to be moving toward the introduction of the first microRNA mimetics using lipid-based formulations (Trang et al., 2011), similar to the administration of siRNAs. Like the siRNA field, there will be multiple flavors of lipid-based formulations to be characterized before efficient delivery is achieved, and each of the formulations will introduce its own set of biologic effects in addition to the microRNA-induced effects. Sorting the microRNA biology from the formulation biology will be another challenge for the technology.
Outlook
The field of microRNA therapeutics is in its infancy and yet already there are promising data in both laboratory animals and in humans. Clinical results with miR-122 inhibition in HCV patients validates the concept that microRNA modulation in humans is possible. The fascinating complexity of microRNA biology offers a wealth of therapeutic opportunities but also poses unique challenges for the development of microRNA modulators as drugs. It is our hope that experimentation to address these challenges will accelerate the advancement of microRNA modulating drugs. With more microRNA therapeutics moving to the clinic, data to address the issues raised herein will be forthcoming, and what is speculation today will be supported or refuted by data to come. That being said, the basic concept of exploiting microRNAs and the evolutionarily conserved pathways that they control should provide for many important disease-modifying therapeutics.
Footnotes
Author Disclosure Statement
A.L.J. holds stock options in Regulus Therapeutics, a microRNA therapeutics company, and is a consultant for miRagen Therapeutics, a company developing microRNA therapeutics for cardiovascular and muscle disease. A.A.L. is employed by miRagen Therapeutics, and holds stock and/or options in Isis Pharmaceuticals, Santaris Pharma A/S, as well as consulting interests with companies working in oligonucleotide therapeutics.