
Abstract

Introduction
Even though the use of aptamers as therapeutic agents is promising, there are challenges that need to be met. One is that the size and polyanionic character of aptamers limits intracellular delivery. However, recent in vivo results on aptamer-mediated specific delivery of an anti-cancer small interfering RNA (siRNA) to prostate cancer cells (Dassie et al., 2009) have not only underlined the potential applicability of aptamers as drug delivery constructs but also give hope for intracellular applications of therapeutic aptamers. Importantly, aptamers, contrary to siRNA and antisense constructs, obviously can be applied against extracellular targets. Size is not only an obstacle for intracellular delivery, but also poses a challenge in manufacturing, as current production technology relies on DNA synthesizers for automated solid support-based synthesis. Another challenge is biodistribution, as unconjugated oligonucleotides are rapidly excreted via the kidneys upon intravenous administration. Conjugation with polyethyene glycol (PEG) units (Veronese and Pasut, 2005) is an approach that has been applied to improve biodistribution of aptamers. The SELEX procedure also can be limiting, as it is time consuming if performed in the standard manual fashion. Automation may in some cases alleviate this challenge (Eulberg et al., 2005; Wochner et al., 2007), and alternative selection/evolution strategies are being developed (Drabovich et al., 2006; Nitsche et al., 2007; Aquino-Jarquin and Toscano-Garibay, 2011; Arnold et al., 2012).
One aptamer has been approved as drug (Pegaptanib) to treat age-related macular degeneration, and others are, or have been, in various stages of clinical development (FAMULOK, 2009; Keefe et al., 2010). In general, the therapeutic candidates have been obtained by post-SELEX modification of aptamers evolved by a full SELEX procedure including 10–20 rounds of selection and enrichment. Post-SELEX modification typically involves truncation into shorter aptamer candidates, conjugation (e.g., PEGylation) for improved biodistribution, and incorporation of chemically modified nucleotides for improved biostability. Post-SELEX chemical modification is necessary, as rather few modified nucleoside triphosphates, for example, 2′-fluoro-RNA, 2′-amino-RNA and 5-substituted pyrimidine nucleoside triphosphates (MAYER, 2009; Lauridsen et al., 2012), are substrates for the polymerase-catalyzed reactions required for efficient SELEX procedures. Post-SELEX modifications are performed in iterative rounds of synthesis and biological evaluation to ensure that modifications are compatible with the desired aptamer properties.
A prominent nucleotide modification in relation to nucleic acid drug discovery is locked nucleic acid (LNA, Fig. 1) (Imanishi and Obika, 2002; Jepsen and Wengel, 2004; Veedu and Wengel, 2010). Incorporation of LNA nucleotides into DNA or RNA strands induces unrivalled increases in duplex thermal stabilities, and LNA phosphorothioate oligonucleotides have shown unique characteristics as single-stranded antisense molecule targeting RNA, for example, messenger RNA or microRNA (Jepsen and Wengel, 2004; Lanford et al., 2010). LNA nucleotides increase nucleolytic stability of oligonucleotides, and their pronounced duplex-stabilizing effect furthermore renders LNA-modified siRNA duplexes highly biostable (Glud et al., 2009).

Chemical structure of DNA, RNA, and locked nucleic acids (LNA).
In our research group, we are currently working on development and evolution of LNA-containing aptamers (Veedu and Wengel, 2009). We want to explore LNA toward a breakthrough within the aptamer field. As mentioned above, despite obvious benefits and two decades of research and development, only relatively few aptamers are currently undergoing clinical development. We therefore think that the need for new approaches and methods to obtain short aptamers with increased biostability, structural stability, and biological function is evident. Post-SELEX LNA modification of aptamers as summarized below have been reported to yield some of these desired properties, but we simultaneously focus on enabling efficient and reliable de novo evolution of LNA aptamers.
LNA-Containing Aptamers by Post-SELEX Modification
LNA in aptamers have so far been reported only by post-SELEX modifications, where LNA nucleotides typically have been placed in the termini or proposed stem regions of already evolved DNA/RNA aptamers. A structure–activity relationship study was carried out for a Tenascin-C binding aptamer, in which unmodified RNA monomers were replaced by 2′-O-methyl-RNA, 2′-fluoro-RNA or LNA monomers. LNA incorporations in one out of three proposed stem regions markedly improved plasma stability while maintaining target binding (Schmidt et al., 2004). Another aptamer selected against the T-cell Leukemia cell line CCRF-CEM was modified with LNA in various positions. LNA in the terminal stem region, along with introduction of a PEG linker in a proposed loop region, allowed for substantial truncation while maintaining target affinity (Shangguan et al., 2007). Similarly, LNA was incorporated into a 21-nucleotide long truncated version of an avidin-binding DNA aptamer; depending on the position of LNA substitution, avidin binding was improved or reduced relative to the all-DNA aptamer (Hernandez et al., 2009). A similar approach was described in a recent patent application where the inventors report increased target binding upon truncation and LNA incorporation in the stem region of a rodent immunoglobulin-G aptamer (Takenaka et al., 2011). A thrombin-binding aptamer was 3′-end capped using the triphosphate derivative of an LNA nucleoside and terminal deoxynucleotidyl transferase. Relative to the non-LNA aptamer, this led to a decrease in the rate of degradation by snake venom phosphodiesterase 1 or human serum (3.6- and 1.5-fold respectively) (Kasahara et al., 2010). An aptamer specific for the B-cell receptor on human lymphoma and leukemia cells was truncated and modified with LNA in the stem region, and also in this case, prolonged serum stability and increased target affinity was achieved (Mallikaratchy et al., 2011). Most recently, a ricin aptamer was modified with LNA in various regions, and in accordance with the above studies, LNA substitutions were tolerated in the stem region, while LNA in loop or bulged regions generally resulted in substantial decreases in affinity (Förster et al., 2012).
In summary, the results obtained with specific LNA substitutions in aptamers derived by post-SELEX modifications point in the direction of increased biostability, increased structural stability, potential for truncation of stem regions, and in some cases increased target affinity. These results underline that LNA may be a key element to realize the full potential of therapeutic aptamers.
LNA-Containing Aptamers by De Novo Evolution
It seems advantageous to be able to directly evolve LNA-containing aptamers in order to most favorably explore the biostability and intramolecular base pairing of short LNA oligonucleotides. Nonetheless, no reports exist on generation of LNA aptamers by in vitro selection, which likely is due to lack of compatibility of LNA nucleotides with the polymerase-catalyzed reactions involved in the necessary SELEX processes. Such an LNA-including SELEX process could involve the steps illustrated in Fig. 2 and summarized below.
Step 1. Library generation. Random pools of LNA oligonucleotides are synthesied by automated procedures using standard phosphoramidite chemistry on a DNA synthesizer. Automated LNA synthesis is known to be compatible with DNA or RNA nucleotides as well as labels and modifiers. LNA phosphoramidite derivatives are commercially available, which makes synthesis of LNA-containing DNA/RNA libraries a relatively straightforward process. Step 2. Selection and partition. The LNA oligonucleotide library is incubated with the specific target of interest, and the most strongly bound sequences are separated. This step is expected to be functional for LNA-including libraries. Step 3. Amplification. The selected LNA oligonucleotide sequences are amplified by PCR, either directly or indirectly, which thus may include PCR to directly furnish LNA/DNA libraries, PCR to furnish DNA-DNA libraries followed by repeated primer extension to regenerate LNA-DNA libraries, or reverse transcription followed by transcription when using LNA/RNA libraries. These steps require compatibility of LNA nucleoside triphosphates and LNA-containing templates with polymerase activity (primer extension, PCR, reverse transcription, and/or in vitro transcription). The following DNA polymerases have been identified as able to incorporate LNA triphosphates into DNA strands opposite to DNA and LNA nucleotides of a template strand: 9° North (Veedu et al., 2007c; Veedu et al., 2008), Phusion high-fidelity (Veedu et al., 2007a; Veedu et al., 2007b; Kuwahara et al., 2008), Taq (Kuwahara et al., 2008), Vent (exo-) (Kuwahara et al., 2008), and KOD (Kuwahara et al., 2008; Veedu et al., 2009; Veedu et al., 2010). Further, transcription using T7 RNA polymerase has been shown to work with LNA nucleotides (Veedu et al., 2008), and the first report on successful reverse transcription with LNA nucleotides has been published (Crouzier et al., 2012). Primer extension reactions are typically effective when using LNA nucleotides and LNA-containing templates, and up to 21 consecutive LNA nucleotides have been incorporated using KOD DNA polymerase (Veedu et al., 2010). Generally, KOD DNA polymerase is a good choice for PCR of LNA-containing DNA duplexes, but results on amplification in our preliminary experiments indicate a limitation in the number of LNA nucleotides that can be included in the substrate if the goal is to amplify directly the LNA-containing PCR substrate. Furthermore, there seems to be a variation in PCR efficiency among different sequences (Veedu et al., 2008; Wengel et al., unpublished data). Engineered polymerases may offer alternative opportunities for enzymatic synthesis of LNA-containing oligonucleotides, as shown recently (Pinheiro et al., 2012), but commercially available polymerases are convenient first choice options. Step 4. Isolation and characterization. After the last round of selection and amplification, the LNA aptamers are converted into double-stranded DNA in order to be cloned and sequenced. Upon analysis of consensus sequence fragments, promising aptamer candidates are resynthesized for binding analysis and/or biological studies.
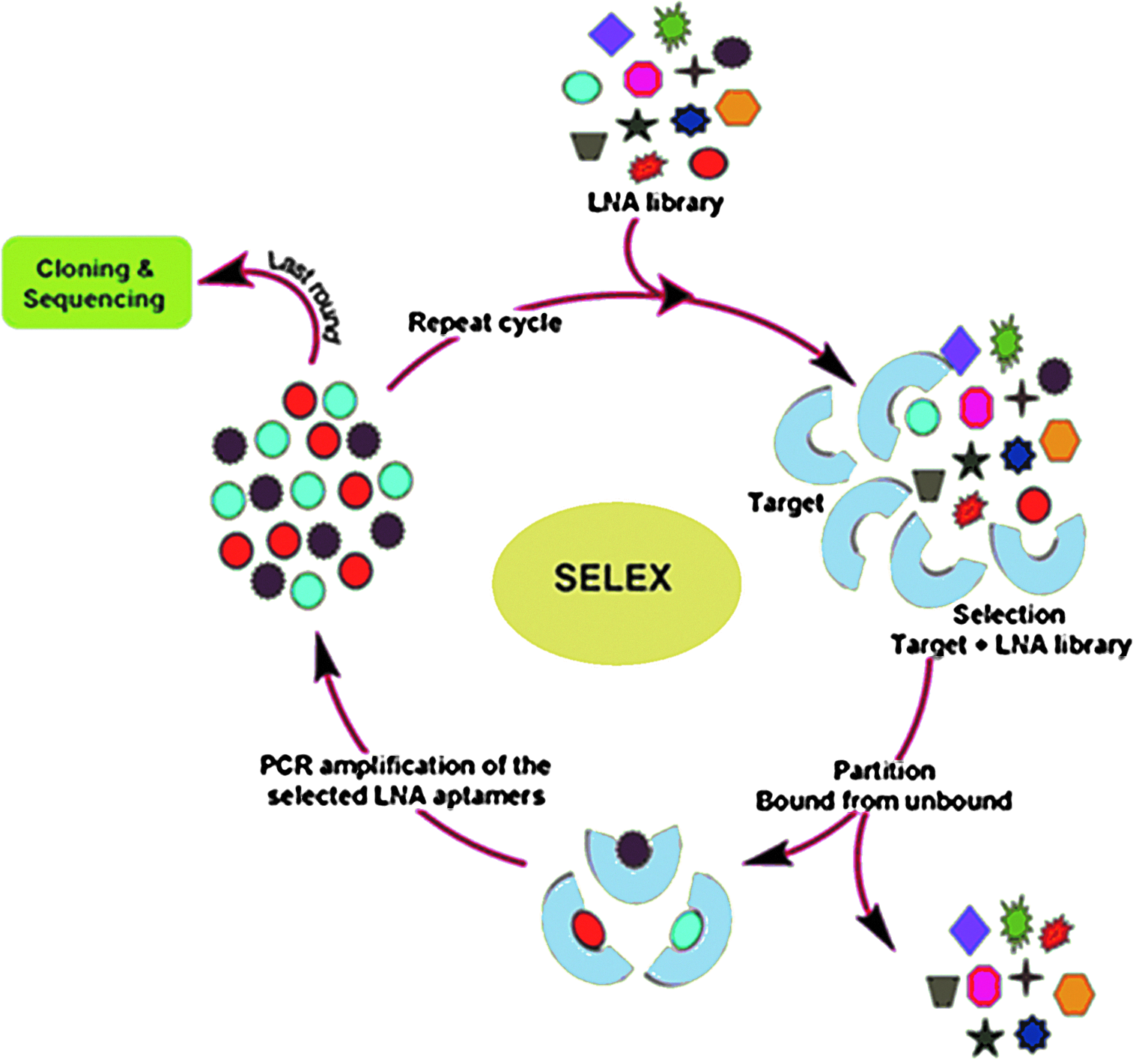
Schematic illustration of an example LNA aptamer generation by systematic evolution of ligands by exponential enrichment (SELEX). Reprinted with permission from Veedu and Wengel, 2009.
Future Prospects
We are currently working on in vitro and in vivo selection of LNA containing aptamers, where LNA monomers are situated in either the 5′-primer region or in the random region. We are exploring strategies involving both direct PCR of LNA-containing binders and indirect amplification via double-stranded DNA. It is our expectation that experimental approaches will soon be established that enable evolution of LNA-containing aptamers, either embarking on commercial or engineered polymerases. Such procedures will, together with post-SELEX modifications, allow for evaluation of LNA as a constituent of aptamers. Encouragingly, aptamers based on hexitol nucleic acid (HNA; a nucleic acid analogue with base-pairing strength in between DNA and LNA) were recently evolved (Pinheiro et al., 2012) as a first step towards a new era in aptamer research and development.
Footnotes
Acknowledgments
The authors would like to acknowledge the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC Grant agreement No. 268776.
Author Disclosure Statement
No competing financial interests exist.