
Abstract
Large-scale RNA interference experiments, especially the ones based on short-interfering RNA (siRNA) technology have become increasingly popular over the past years. Obviously, the sequence (and structure) of the corresponding siRNA is a key factor in obtaining reliable results in these large-scale studies, and the companies use a variety of algorithms to design them. Design tools have been developed based on experimental data to increase the knockdown efficiency of siRNAs. Nevertheless, as the genome annotations are still continuously changing, siRNAs may become obsolete, so siRNA reagents should be periodically re-annotated according the latest version of sequence database. In this article, existing siRNA design algorithms, design parameters, and siRNAs are evaluated. A new approach for systematic analysis and re-annotation of siRNAs libraries produced in the last decade is introduced here.
Introduction
Due to technical/practical reasons, there is always a time-gap between the design and the actual production/delivery of the reagents. Also, because of the relatively high costs the researchers use a certain library for years annotated in the year of delivery. Consequently, as genome annotations are still changing, there is a need to reanalyze the libraries with the latest National Center for Biotechnology Information (NCBI) reference sequences (RefSeq) (Pruitt et al., 2007). The quality of reagents and reagent-to-gene-model linkages/relations must be reanalyzed at regular intervals. Reagent designs with old RefSeq versions may have a lack of homology to any known mammalian gene in the latest RefSeq version. Based on the new information, constructs may also match to a different mRNA transcript sequence. Ultimately, RNAi reagent quality can serve to characterize unknown proteins and provide guideposts for follow-up analysis. Analysis of existing libraries with the latest annotation and reagent knockdown specificity are essential for cross-correlating phenotypic experiment information. At the same time, a comparison across multiple experiments can also be used to evaluate and confirm the reliability of a particular large-scale RNA interference (RNAi) reagent (i.e., side effects). This paper proposes an approach to deal with the problem of the outdated siRNA annotation and design quality by using on-target analysis. The result of our analysis will show that, in general, most design algorithms still output quite good collections of effective siRNAs. Nevertheless, these libraries contain a large number of siRNAs which must be re-annotated or discarded from experimental data.
Methods
The aim of the complementary search is to determine whether there exists a complementary region between the selected siRNA sequences and the messenger RNA (mRNA). Many different sequence alignment algorithms could be used for such a complementary search, but they are they are not necessarily optimal for this purpose by default. In our approach we used/combined 3 different strategies to find nearly exact complementary regions as well as small local complementarities. Our complementary search was based on Basic Local Alignment Search Tool and Smith-Waterman algorithms. Because of the runtime problem when performing a local alignment with the Smith-Waterman algorithm, a third variant, Seed-Motif-Search combined with the Smith-Waterman algorithm, is introduced here. In this variant, an initial step reduces the length of the mRNA sequences to enable the use of a local alignment algorithm. This reduction is made because the seed region (2–7 position on siRNA antisense sequence) of the siRNA seems to play a significant role in causing non-target effects. At the beginning, all occurrences of the seed motif of every siRNA are localized in the genes. After detecting this small region, a sequence of ∼50 nt around this seed motif is cut out in the mRNA. Thus, as a result of this first step, a huge number of sequences of ∼50 nt in length are obtained containing the seed region of each siRNA. Due to the small length of the sequences it is now possible to perform a local alignment with the Smith-Waterman algorithm. The advantage of the Seed-Motif-Search for on-target analysis is that it limits the results to those genes that perfectly match with the seed region of the siRNA.
To asses design parameters of commercially available libraries, an on-target analysis was performed using design rules we derived from various sources to rationalize the redesign process. Existing design rules were assembled from various algorithms (Elbashir et al., 2001; Amarzguioui et al., 2004; Reynolds et al., 2004; The siRNA user guide, 2004; Ui-Tei, et al. 2004.
A collection of the most-applied parameters in currently available design algorithms is shown in Table 1. The parameters P2–P5, P7–P10, P12, P17, and P18 enhance the siRNA efficacy, whereas P1, P6, P11, and P13–P16 reduce the efficacy of siRNA. Design parameters were computed using rules we derived from various sources to rationalize the original process and to understand design algorithms schemas (redesign). The A or T at position 10 (P4) is at the cleavage site and may promote catalytic RISC-mediated passenger strand and substrate cleavage. In addition, moderately low GC content (30%–55%) contributes to efficiency (P12). Some studies have shown that the presence of elevated GC content at the 5′ end of the siRNA target sequence improves the siRNA efficiency (Elbashir et al., 2001) (P11). Analyses based on the individual positions of the siRNA target sequence have shown that the presence of bases G/C at position 1 (P2), A/T at positions 16–19 (P7–P10), A at position 3 (P3), and A/T at positions 10 and 13 (P4, P5) of the sense strands positively affect the siRNA efficiency, whereas the presence of bases G/C at position 19 (P11), G at position 13 (P6), and A/T at position 1 (P1) negatively affect the siRNA efficiency (Elbashir et al., 2001; Amarzguioui et al., 2004; Reynolds et al., 2004; Ui-Tei, et al. 2004). Target position is the most reliable mRNA sequence information available (P18). Because the coding sequence is the most reliable mRNA sequence information available, it is commonly targeted. The untranslated regions (UTRs) are generally less well characterized but can also be targeted with similar gene-knockdown efficiency (McManus, et al., 2002; Harborth, et al., 2003; Hsieh, et al., 2004).
The parameters are classified into 2 categories according to their significance for efficiency: increase=enhance the siRNA efficacy, and reduce=suppress the efficacy of siRNA.
Long double-stranded RNAs that have regions of low complexity [for example, CA[ATCG] repeats (P13) or simple nucleotide repeats (P14)] can exert unspecific and cytotoxic effects (P12). It has been reported that siRNA duplexes can activate innate immune responses by interacting with certain toll-like receptors on the cell surface or in the endosomes (Hornung, et al., 2005; Judge, et al., 2005). The invoking of these responses requires the absence of specific motifs, 5′-TGTGT-3′ or 5′-GTCCTTCAA-3′, in the guide strand of siRNA duplexes (P15, P16).
The 18 design criteria were tested and weights were calculated using the training dataset for each of them in order to build categories. The different weights of all the criteria were computed with the Konstanz Information Mine (KNIME) (Berthold, et al., 2007) “sum squared error” performance node to determine the best fit of the data using a second-order polynomial fit. The best fit weights minimizing the sum squared error function were computed 140 times after randomizing the order in which the design parameters were considered, and the average weight for each criterion was computed. The scores obtained for each siRNA were then normalized in the range of 0–100 using the newly introduced method “Design Ratio”:
where Wi are the weights obtained by each of the 18 criteria (Pi; summarized in Table 1). DRsirna is a ratio obtained by the siRNA sequence after normalization of the raw score used for siRNA specificity ranking.
The specificity of an siRNA construct is a crucial factor in any silencing experiment (Semizarov, et al., 2003). Protein expression silencing through the RNAi machinery works perfectly if the siRNA is totally complementary to its target mRNA. It is well known that single nucleotide mismatches between the siRNA and the target mRNA decrease the rate of mRNA degradation (Haley et al., 2004). The different companies' algorithms for generating the best siRNA sequence typically take this into account and check and exclude siRNA sequences that have total complementarity to other than the target mRNA. Nevertheless, with the evolution of gene annotation and new releases of the RefSeq database (Pruitt et al., 2007), already designed siRNAs today may or may not match to new mRNA transcript sequences. Hence, existing libraries need to be regularly analyzed for their specificity based on sequence-dependent analysis. We call this process as “on-target analysis.” On-target analysis uses the latest reagent annotation and calculates the related specificity based on a complementary search algorithm against latest NCBI databases (Fig. 1). We performed on-target analysis using a customized design/evaluation pipeline, High Content Data Chain (HCDC)-KNIME.
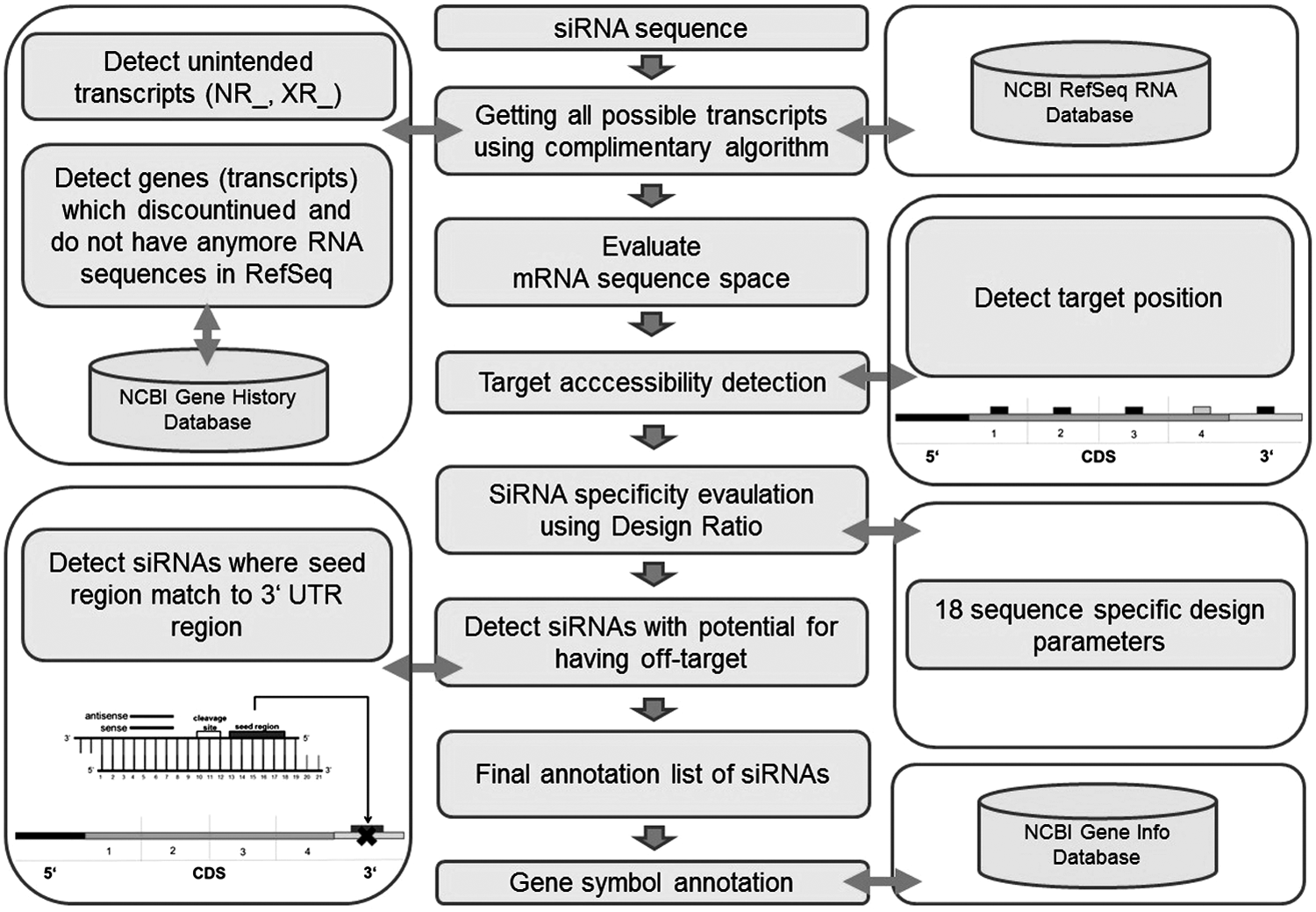
Flow chart of “on-target analysis” approach. On-target analysis takes a small interfering RNA (siRNA) sequence; gets the messenger RNA (mRNA) sequence from the RefSeq database; finds out if this messenger RNA (mRNA) has other transcript(s); performs multiple sequence alignment with the transcripts, if any, and takes the consensus un-gapped sequence; labels unintended transcripts (NR, non-coding transcripts including structural RNAs; XR, non-coding predicted transcripts; or discontinued in history); performs target accessibility evaluation; predicts siRNA efficiency using the implemented design ratio; labels the candidates that pass the threshold assigned for each of the 18 parameters used; labels siRNA targeting 3′untargeted regions (UTRs) as off-targeted mRNA, either complete or on seed homology; detects siRNA that target 3′ on seed region; and updates gene symbols based on the latest Gene Info Database.
Results
Design parameters
In order to test the validity of presented siRNA design parameters, 3 human genome-wide libraries from commercial suppliers were evaluated. This enabled the analysis of 26,393 siRNAs in the case of the Applied Bioscience (Ambion) library, 71,593 in the Qiagen library, and 71,764 siRNAs in the Thermo Scientific (Dharmacon) library (Fig. 2). In this study we report that currently used rules in design algorithms are not sufficient to accurately distinguish between potent and weak RNAi triggers. Currently, rule-based siRNA design tools are often prone to low sensitivity. One strategy for countering the low sensitivity problem is to consider all existing rules, rather than individual rules, to select most potential siRNA. The properties given in common principles are not exclusive for effective siRNAs.
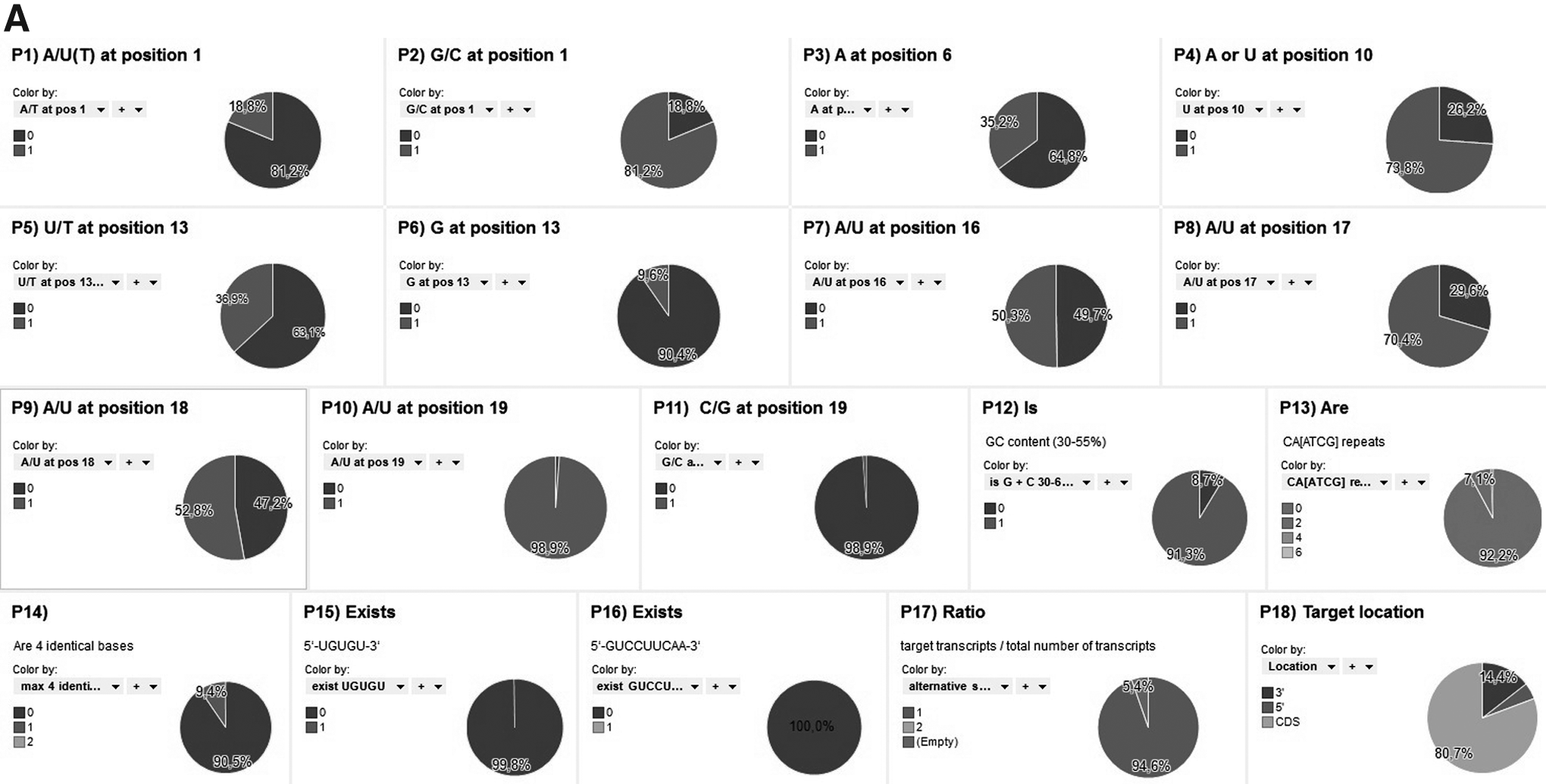
Statistics for 18 design parameters for human genome–wide libraries


In order to calculate the correlation of the above-mentioned parameters, it was important to introduce an efficiency category for each parameter. Two efficiency categories for all of the criteria were generated: increase and reduce (Table 1). After creating appropriate groups it was realized that, for example, A/T at position 1 of the sense strand, G at position 13 of the sense strand, and G/C at position 19 of the sense strand did not significantly affect the siRNA efficiency, as they acquired the lowest weights. The AAAA motif or the TTTT motif has the tendency to cause the premature termination of transcription of siRNAs from the RNA polymerase 3 promoter (Geiduscheck, et al., 2001). Four identical bases in the siRNA sequence (GGGG, CCCC, TTTT, AAAA; parameter P14) can cause potential nonspecific effects through its interaction with heparin-binding proteins. In addition, siRNA sequences should not contain specific motifs [5′-TGTGT-3′ (P15) or 5′-GTCCTTCAA-3′ (P16)] in the guide strand of siRNA duplexes. in existing libraries, we have exploited about 2% of siRNAs from Qiagen, 1.8% of siRNAs from Dharmacon, and 0% of siRNAs from Ambion having in sequence 5′-TGTGT-3′ motifs. Fortunately, in all libraries there are no constructs having in sequence 5′-GTCCTTCAA-3′ motif. Of the Qiagen, Dharmacon, and Ambion siRNAs having in sequence 4 identical bases (parameter P14) 0.0%, 5.7%, and 9.2%, respectively, have been calculated. Thus, the amount of inefficient design siRNAs in libraries is significant, and such siRNA should be labeled as less effective.
It has been reported that target accessibility can improve the target knockdown level significantly. Target site identification was tested by full complementarity of 3 libraries. Our report shows that 59.6% of siRNAs from Qiagen, 63.8% of siRNAs from Dharmacon, and 80.7% of siRNAs from Ambion mapped to the <Please define CDSCDS, whereas, respectively, 35.7% of Qiagen, 36.2% of Dhramcon, and 14.4% of Ambion matched a 3′ UTR. On average at least one siRNA from all for each target gene should match on the 3′ UTR of the mRNA. Targeting 3′ UTRs allows a rescue experiment of expression, which can then be achieved by simply expressing (plasmid/vector) a form of the mRNA lacking its normal 3′ UTR. SiRNAs may cause unspecific gene silencing through perfect sequence homologies to the siRNA matches to the transcript 5′ UTRs. Although it is not desired, 3 libraries still contain siRNAs with almost perfect matches to 5′ UTR (Qiagen, 4.7%; Dharmacon, 0.0%; and Ambion, 4.9%).
An important parameter is the alternative splicing ratio (P17), as the entire gene transcripts should be assigned for targeting, and only the conserved regions between multiple transcripts should be targeted. One mismatch existing between alternative transcripts and siRNA may dramatically affect siRNA efficiency. A comparison between the libraries showed how the design strategies considered alternative splicing. Of the Qiagen, Dharmacon, and Ambion siRNA ratios (parameter P17, siRNA target transcripts/total number of transcripts), 95.5%, 98.3%, and 94.6%, respectively, have been calculated with a ratio of 1.
Analyses based on the individual positions of the siRNA target sequence have shown that the presence of bases G/C at position 1 (Qiagen, 39.3%; Dharmacon, 28.3%, Ambion, 10.9%); A at position 6 (Qiagen, 24.6%; Dharmacon, 33.2%; Ambion, 35.2%); A or T (A is more common) at position 10 (Qiagen, 50.8%; Dharmacon, 62.4%; Ambion,
siRNA, small interfering RNA.
In our study we propose an approach to designing siRNA, passing most of the design criteria used in design algorithms. We suggest that in the siRNA, a presence of bases G/C at position 1 (P2), base A at position 6 (P2), bases A/T at position 10 (P4), and base T at position 13 (P5). For positions 16–18, we propose combination of A/T residues (P7, P8, P9, and P10) that increase efficiency, avoiding G/C at position 1 (P11), and to avoid 3 or 4 identical bases in the siRNA sequence (P14). In order to get more potent siRNA, we suggest mutation on positions 2–5, 7–9, 11, 12, 14, and 15 (sense strand) with all four nucleotides of a siRNA. Efficacies of these mutated siRNA combinations can be control using desiRm model (www.imtech.res.in/raghava/desirm/) (Ahmed et al., 2011). It should be also considered that in those positions, moderately low GC content (30%–55%) should be achieved. In addition, siRNAs with (A+T)-rich seed regions have more 3′ UTR binding sites, a consequence of the (A+T)-rich nucleotide composition of 3′ UTRs is dilution effect on each target message. Indeed, A+T can be manipulated to titrate siRNAs away from their normal targets, and natural A+T has been proposed to influence siRNA off-targeting (Anderson et al., 2008; Arvey et al., 2010). That is why it is important to introduce G/C content into seed region (position 2–7 antisense strand).
Library update with latest gene and transcript annotation
An important challenge remains the systematic analysis and annotation of siRNAs reagents according to latest versions of genome databases. The on-target analysis results of commercially available human genome wide RNAi (2 genome wide) libraries designed and delivered in different time were calculated and presented on Fig. 3.

Statistics from on-target analysis for 2 genome-wide libraries: Qiagen (library designed and delivered in 2007) and Dharmacon (library design and delivered in 2010).
As a result of on-target analysis, we classified siRNAs in the following categories:
• Wrong design: if oligonucleotide does not target the gene annotated by siRNA supplier, but rather targets a different gene.
• Is pseudogene: if target gene is a pseudogene.
• Real off-targets: if in addition to the originally designed target gene, oligonucleotide also targets another gene.
• Number of transcripts: provides information about the number of transcripts of the targeted gene.
• Position on mRNA: displays target site identification as a position of siRNA on mRNA sequence.
• Non-target: is true for siRNA if there is no target gene with sequence complementarity in entire genome for designed reagent.
• Location (target site): indicates a target location (CDS, 5′, 2′) and a specific region on CDS (divided in 4 sectors).
• Number of unintended transcripts (XM—mRNA of under curation; NR—non-coding transcripts including structural RNAs; transcribed pseudogenes; and others).
• New gene symbol for GeneID.
• Design ratio (see methods section).
On an average, approximately 7% of siRNAs per library were labeled as non-target reagents, 1% as wrong design, 3% as off-target, and 1% as a pseudogene; 55% target 1 mRNA transcript, 17% target 2 mRNA transcripts, 7% target 3 mRNA transcripts, and 21% target more than 3 transcripts. We have exploited in existing libraries approximately 5,467 of siRNAs from Qiagen and 3,290 of siRNAs from Dharmacon targeting unintended NR transcripts; 1,090 of siRNAs from Qiagen and 192 of siRNAs from Dharmacon targeting unintended XM transcripts; and 11,0424 of siRNAs from Qiagen and 110,468 of siRNAs from Dharmacon targeting intended NM transcripts. Our report shows that 37% of siRNAs from Dharmacon and 6.2% of siRNAs from Qiagen for the same GeneID received a new gene symbol. The latest GeneID to GeneSymbol mapping was received by querying the NCBI Gene Info Database. Figure 4 demonstrates on-target analysis output as a list of 3 selected siRNAs for each library, their sequences, and on-target analysis results. Such a table should be incorporated into experimental data from large-scale siRNA experiments in order to verify more precisely the selection of potential “hit” candidates.
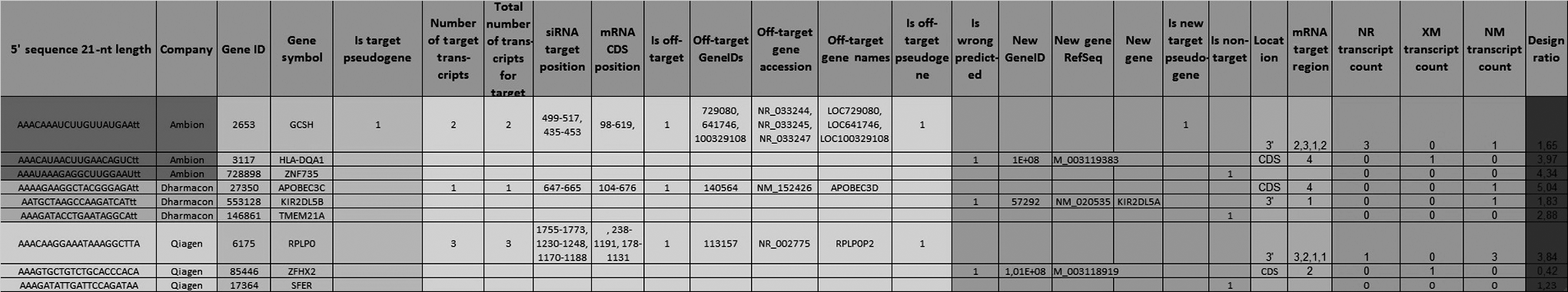
Example of siRNAs and their on-target analysis results are shown from Qiagen, Ambion, and Dharmacon libraries, respectively, and analyzed with RefSeq 2012. The columns can be classified into 8 functional groups that contain information about original target, transcripts, off-targets, wrong design, non-target, location on mRNA, number of different transcripts, and design ratio.
Experimental validation
To validate the design parameters, we used 2 RNAi cell-based screening data sets from our previously published experiments (Collinet et al., 2010; Wild et al., 2010).
Testing with Collinet data
The goal of the project was to study the endocytosis process fulfilling many cellular and developmental functions (Collinet et al., 2010). A new strategy to phenotypically profile the human genome with respect to transferrin (TF) and epidermal growth factor (EGF) endocytosis by combining RNAi, automated high-resolution confocal microscopy, quantitative multiparametric image analysis, and high-performance computing has been developed. Several novel components of endocytic trafficking, including genes implicated in human diseases, have been invented. Endocytosis visualization of fluorescent TF and EGF in HeLa cells was applied. Three genomic RNAi libraries, 2 commercial siRNA libraries (Ambion Silencer Genome Wide siRNA Library V.3 and Qiagen Human Whole Genome siRNA Library V.1) and a custom-made endoribonuclease-prepared siRNAs (esiRNA) library were screened.
The reference table (Collinet et al., 2010; Supplementary Tables 1, 2; Supplementary Data are available online at www.liebertpub.com/nat) results include the sequences of all si/esiRNAs targeting the 4609 positive genes in the screen. A list of 4,609 genes—3,804 with a “strong” phenotype and 805 with a “mild” phenotype—was compiled: 84% reproducibility was observed for strong and 72.5% for mild phenotypes. In the table from the 17,867 Qiagen siRNAs targeting 4,609 positive genes in the endocytosis screen, we identified 277 siRNAs that were labeled as perfectly designed (Fig. 5; Supplementary Table 1) and 6 as wrong designed (Supplementary Table 2). Zerial group (www.mpi-cbg.de/research/research-groups/marino-zerial.html) identified 14 phenotypic cluster groups stable over a wide range of the algorithm parameters, indicating that the endocytic system is under constraints and changes between a discrete set of possible states in response to perturbations. The groups of phenotypes include selective up-regulation of TF endocytosis; selective down-regulation of TF endocytosis; and 12 specific effects on subcellular localization: (1) endosomes appear clustered in the cell centre, (2) specific effect on subcellular localization: endosomes appear dispersed in the cell periphery, (3) opposite effects on EGF and TF endocytosis: EGF endocytosis is increased and TF endocytosis is decreased, (4) opposite effects on EGF and TF endocytosis: EGF endocytosis is decreased and TF endocytosis is increased, (5) effects on endocytosis of both markers: increased EGF and TF endocytosis, (6) effects on endocytosis of both markers: decreased EGF and TF endocytosis, (7) selective up-regulation of EGF endocytosis, (8) selective down-regulation of EGF endocytosis, (9) selective up-regulation of EGF endocytosis with accumulation of endosomes in cell centre, (10) reduced TF endocytosis with endosomes accumulated in the cell centre, (11) selective increase in EGF endosomes number and elongation, and (12) increase in elongation of TF endosomes with mild increase of TF endocytosis. From the selected list of strong oligos (perfect design), CDO1 gave phenotype specific effect on subcellular localization: endosomes appear clustered in the cell centre, with hit category “strong;” and the oligo for ARG3 (ARG1) gave the phenotype selective up-regulation of TF endocytosis, with hit category “strong” (Fig. 5). Selected siRNAs for gene CDO1 had very high DRsirna (4.59) and for gene ARG3, DRsirna=5.78.
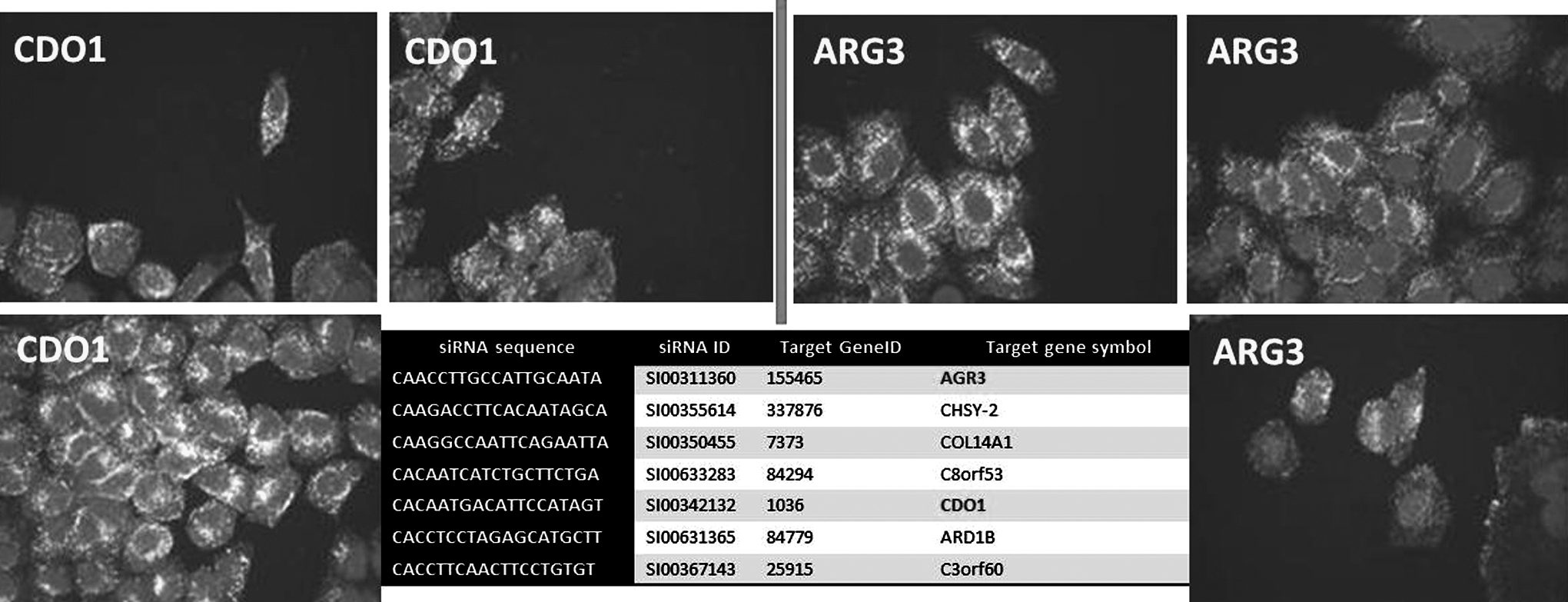
Selected list of optimal designed siRNA and their phenotypic results in Zerial screen. Two optimal designed siNRAs for gene ARG3 and CDO1 gave very strong hits in Zerial screen: CDO1: phenotype, specific effect on subcellular localization: endosomes appear clustered in the cell centre; hit category: STRONG and ARG3 (ARG1): phenotype, selective up-regulation of TF endocytosis; hit category: STRONG.
For comparison with wrong design oligos, we selected 3 siRNAs as an example and checked their target gene phenotypes (Table 3). The wrong design oligos have very low DRsirna and belong to the group of siRNAs that did not give a hit in the screen.
DRsirna, design ratio for siRNA; EGF, epidermal growth factor; TF, transferrin.
Testing with wild data
We used a biogenesis kinase screen to examine whether perfectly designed oligos are part of a hit list. To monitor 60S biogenesis (Wild et al., 2010) we established a HeLa cell line carrying an inducible copy of the ribosomal reporter protein Rpl29-GFP. Rpl29-green fluorescent protein (GFP) is efficiently incorporated into 60S subunits, and the GFP readout faithfully illustrates the Crm1 dependency of 60S export.
A HeLa cell line bearing a tetracycline-inducible Rpl29-GFP gene is used in this assay. Upon induction of Rpl29-GFP expression, Rpl29-GFP is incorporated into newly synthesized ribosomal particles and within the time of induction used in this experiment (14 hours), a nucleolar and cytoplasmic signal is visible (see images of negative control conditions in Fig. 6). When nuclear 60S biogenesis events are perturbed, Rpl29-GFP accumulates in the nucleus. A custom-assembled siRNA library was synthesized by Qiagen. SiRNAs (10 μL of a 100 nM stock in reduced serum media-minimum essential media) were added to the transfection reagent [0.0625 μL Oligofectamin (Invitrogen) in 20 μL OptiMEM] and incubated at room temperature for 30 minutes. Then, 70 μL of cells (1,750 HeLa Rpl29-GFP cells) were added to each well and incubated at 37°C and 5% CO2. Fifty-eight hours after transfection, cells were induced with tetracycline (final concentration of 125 ng/mL) for 14 hours and subsequently incubated for 6 hours in tetracycline-free medium. Thereafter, cells were fixed with 4% paraformaldehyde and stained with Hoechst. Nine images were taken using a 20× objective on a BD pathway 855 microscope. AllStars (Qiagen) was included as a negative control. siRNA targeting Crm1 (Xpo1) was present as a positive control. Two phenotypes have been distinguished in the screen using the Rps2-YFP reporter, reflecting early (nucleolar) and late Rps2-GFP (nucleoplasmic) defects in nuclear pre-40S maturation. Rpl29-GFP is efficiently incorporated into 60S subunits, and the GFP readout faithfully illustrates the Crm1 dependency of 60S export.
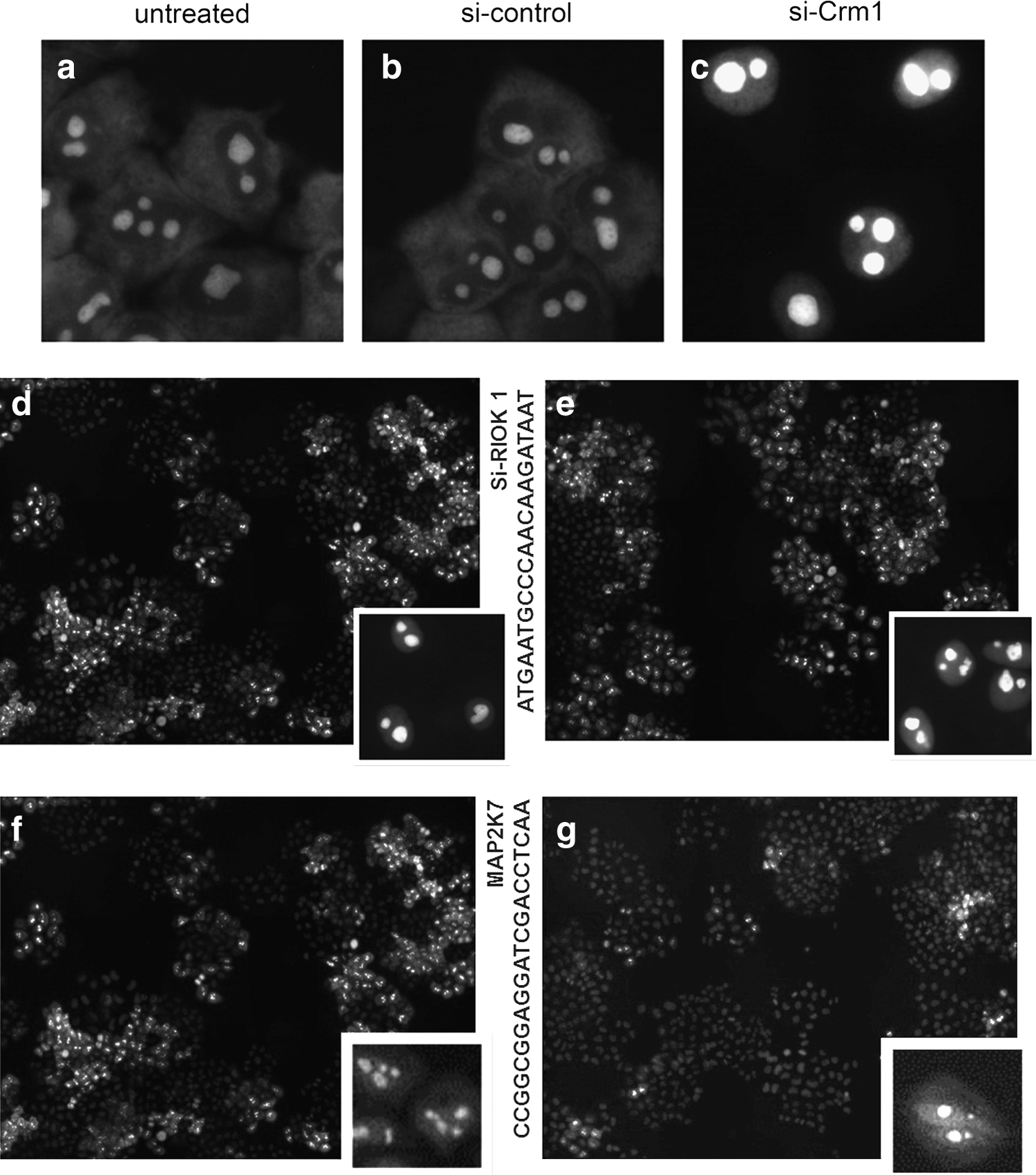
Behavior of perfectly designed siRNAs targeting CRM1, RIOK1, and MAP2K7 in HeLa cells.
As shown in Supplementary Tables 3 and 4, from 2,980 (4 oligo) siRNAs there are 62 sequences defined in the Wild screen as hits, which we classified as perfectly designed (Fig. 6; Supplementary Table 3) and 8 with low ratio design, which did not show any efficiency in the screen (Supplementary Table 4). Figure 6a, b, and c show epifluorescence microscopy images showing Rpl29-GFP readout of untreated, siRNA control, and siRNA-CRM1 treated HeLa cells. Epifluorescence microscopy images in Fig. 6d and e show Rpl29-GFP hit readout of siRNA-RIOK1, indicating phenotype as untreated treated HeLa cells. Epifluorescence microscopy images in Fig. 6f and g show Rpl29-GFP readout of siRNA-MAP2K7 (Rpl29-GFP phenotype) treated HeLa cells.
Discussion
SiRNA design tools based on selected parameters are often prone to low sensitivity (i.e., they tend to produce rather many siRNAs very often with low quality for a given gene). One strategy for countering the low sensitivity problem is to organize the design rules into disjunctive sets and use the rule sets rather than individual rules. We have evaluated 3 existing libraries (Ambion, Dharmacon, and Qiagen) based on common design rules. The result shows that, in general, most of the design algorithms have implemented the most important design rules but still output quite a number of weak designed constructs. Finally, while considering steps involved in the RNAi pathway, it clearly appears that other phenomena, such as mRNA target location, have to be considered into a strategy for fine-tuned design algorithms.
The on-target analysis demonstrated that quality control at the level of sequence mapping is crucial for the interpretation of large-scale RNAi experiments. An important step in every high throughout experiment is a systematic analysis and re-annotation of siRNA reagents according to the latest versions of transcripts and genome databases. It is thus crucial to improve annotation and design of RNAi libraries showing better specificity and efficiency and reducing unintended off-target effects.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.