
Abstract
To treat urethral strictures of the lower urinary tract, urethrotomy is the method of choice. But this minimally invasive method suffers from poor outcome rates and leads often to restenosis of the urinary tract because of hyper-proliferating fibroblasts. Our aim is to minimize the proliferation of excessive tissue due to a new minimal invasive therapeutic approach. As an appropriate model, we isolated fibroblasts from different benign prostatic hyperplasia patients and transfected them with small interfering RNA (siRNA) against the transcription factor serum response factor (SRF), a key factor for cell cycle regulation and apoptosis. The resulting knockdown of SRF was examined on the messenger RNA level by quantitative real-time polymerase chain reaction and on the protein level by western blot. The correlation of SRF silencing and impact on cell proliferation was examined by xCELLigence, 5-bromo-2′-deoxiuridine proliferation assay, total cell counts, and senescence assay. The transfection of primary prostatic fibroblasts with SRFsiRNA revealed specific and significant knockdown of SRF, leading to significant inhibition of proliferation after the second transfection, which was revealed by proliferation assay and total cell number. The results of this study indicate a substantial role of SRF in prostatic fibroblasts and we suggest that SRF silencing might be used for the treatment of urethral strictures to achieve a durably patent urethra.
Introduction
One possible approach would be the use of the RNA interference (RNAi) mechanism. Recently, Andrew Z. Fire and Craig Mello were awarded the Nobel Prize in medicine and physiology for the discovery of the RNAi pathway (Fire et al., 1998). RNAi has been shown to efficiently silence genes in a variety of mammalian cells, when small interfering RNA (siRNA) duplexes are delivered into cells (Elbashir et al., 2001). The incorporation of the siRNA into the RNA-induced silencing complex, leads to degradation of the complementary messenger RNA (mRNA), and results in a decrease of the corresponding protein amount (Hammond et al., 2000).
One possible target for inhibiting cell proliferation of smooth muscle or HepG2 and JHH6 hepatocellular carcinoma cells is already described by Werth et al. and Farra et al. respectively (Farra et al., 2010; Werth et al., 2010). They found unrecoverable senescence or induced apoptosis after delivery of siRNA against serum response factor (SRF). SRF is a transcription factor of the MADS box family (TREISMAN, 1986; Shore and Sharrocks, 1995) and is involved in various cellular processes such as expression of immediate-early genes and tissue-specific genes, cell proliferation, differentiation, and apoptosis (Bertolotto et al., 2000; Camoretti-Mercado et al., 2000; Ding et al., 2001; Schratt et al., 2001; Zhang et al., 2001). Figure 1 explains the possible therapeutic application of siRNAs to treat urethral strictures.
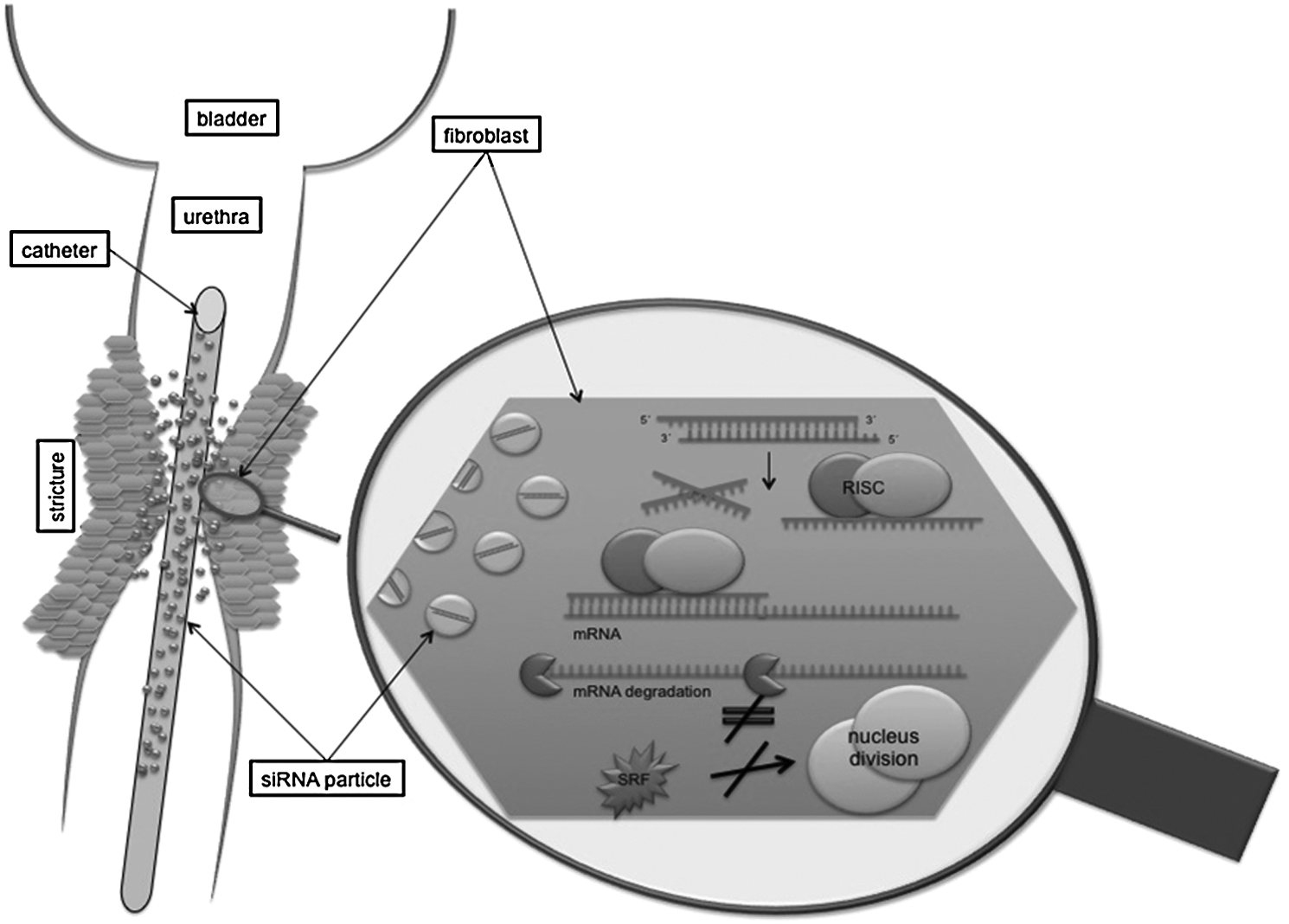
Possible application of the RNA interference (RNAi) mechanism with serum response factor small interfering RNA (SRFsiRNA) immobilization on a catheter to prevent stenosis. siRNA is delivered into the cell by endocytosis; afterward siRNA is introduced into the RNA induced silencing complex, and antisense strand mediates binding to complementary mRNA. Thereby messenger RNA (mRNA) gets degraded and corresponding SRF protein is not built. The depletion of SRF leads to inhibited cell division and therefore to reduced excessive tissue proliferation.
Due to the fact that fibroblasts play a major role in urethral stricture development, we decided to isolate primary fibroblasts from patients suffering from benign prostatic hyperplasia, as it is known that fibroblast proliferation is 37-fold higher in benign prostatic hyperplasia patients compared to healthy patients (Claus et al., 1993). The use of fast duplicating primary fibroblasts demonstrates an adequate model for the examination of siRNA silencing and the impact on cell proliferation. The isolation of fibroblasts was controlled by their cluster of differentiation (CD)90-thymocyte differentiation antigen 1 (Thy-1) expression, a glycophosphatidylinositol-anchored glycoprotein of the immunoglobulin superfamily. The antibody clone AS02 recognizes only fibroblasts, whereas other CD90 antibody clones recognize generally thymocytes, neurons, or mesenchymal stem cells. We analyzed the influence of SRFsiRNA transfection in primary fibroblasts in matters of cell number, cell cycle, and senescence induction. Our study proves helpful for developing new strategies to decrease the risk of urethral stricture restenosis after surgical or traumatic intervention in the urethra and thus greatly improving long-term outcome of urinary tract patency.
Materials and Methods
Isolation, cultivation, and characterization of prostatic fibroblasts
Human primary fibroblasts were isolated from prostatic tissue obtained from patients undergoing transurethral resection of the prostate. All human subjects provided written informed consent with guarantees of confidentiality, and the study was approved by the Clinical Ethics Committee of the University of Tuebingen. Isolation of human primary fibroblasts was performed using the following method: Prostatic tissue was reduced to small pieces and placed in one well of a 6-well plate with Dulbecco's modified Eagle's medium (DMEM)–high glucose medium (PAA Laboratories GmbH, Pasching, Austria) supplemented with 2 mM
Flow cytometry
Isolated cells were examined for CD90 expression with anti-CD90 antibody, clone AS02 supplied from Dianova (Hamburg, Germany). Fibroblasts were stained for 30 minutes with anti-CD90 fluorescein isothiocyanate–labeled antibody or corresponding isotype according to the manufacturer's instructions in 0.5% FCS at 37°C. After cell detachment and fixation, flow cytometry (5,000 cells per measurement) was performed in a FACScan™ from Becton Dickinson GmbH (Heidelberg, Germany) and evaluated with the CellQuestPro software (Becton Dickinson GmbH).
siRNA transfection
For proliferation and senescence assays, 30,000 and 20,000 cells respectively were cultured in 6-well plates 1 day prior transfection. For determining cell numbers, 10,000 cells were cultured in 12-well plates 1 day before transfection. Fibroblasts were transfected with 25 nM SRFsiRNA or si scrambled (siSCR) utilizing the transfection reagent Interferin™ (Polyplus-transfection SA, Illkirch, France). In brief, for 1 well of a 12-well plate with 25 nM siRNA concentration, we diluted 0.4 μL siRNA (20 μM) and 1.17 μL Interferin in 318 μL DMEM–high glucose medium (PAA Laboratories GmbH, Pasching, Austria) without supplements. To form transfection complexes the solutions were incubated for 20 minutes at room temperature. Afterward, the medium of the cells was aspirated and 300 μL of the transfection complexes was pipetted into 1 well of a 12-well plate. Transfection was carried out for 2 hours at 37° C, 5% CO2, and 92% humidity. Afterward, the transfection solution was removed and fresh culture medium was added. For quantitative real-time-polymerase chain reaction (PCR) analysis 40,000 cells per well were cultured in 12-well plates 1 day prior to transfection. On days 1, 4, and 7 transfection was carried out using siRNA concentrations of 1, 10, 25, 50, and 100 nM.
Size and zeta potential characterization of transfection complexes
The size and zeta potential was measured by using a Zetasizer Nano ZS (Malvern Instruments, Malvern, Herrenberg, Germany). For size measurement we used size distribution report for intensity; for zeta potential, the mobility u was converted into zeta potential using the Smoluchowski relation. For analyzing the size and zeta potential, 4.8 μL (20 μM) siRNA and 3.5 μL interferin were pipetted into 1.5 mL ultrapure water and allowed to form transfection complexes for 20 minutes at room temperature before determination of the size and zeta potential.
siRNA sequences
The SRFsiRNA sequence was previously published by Werth et al. with the following sequence: sense, GAUGGAGUUCAUCGACAACAA; antisense, GUUGUCGAUGAACUCCAUCUU (Werth et al., 2010). The nonsilencing siRNA (siSCR) was supplied from Qiagen (Hilden, Germany) and is validated by Affymetrix GeneChip arrays and a variety of cell-based assays and ensures minimal nonspecific effects on gene expression and phenotype.
Quantitative real-time PCR
Total RNA was isolated using AurumTM total RNA mini kit (Bio-Rad Laboratories, Inc., Hercules, CA) and quantified. For reverse transcription, 200 ng of each RNA sample was used with the iScriptTM cDNA Synthesis Kit (Bio-Rad) according to the manufacturer's instructions. All primers were synthesized by Operon Biotechnologies GmbH (Koln, Germany). Primer design was done with the software Primer3 (Rozen and Skaletsky, 2000) and Primer Premier 5 (PREMIER Biosoft International) using IQTMSYBRRGreen Supermix (Bio-Rad). The following primer sequences were used for SRF: (F)5′-AGTGCAGGCCATTCAAGT-3′ (R)5′-ACGGATGACGTCATGATGGTG-3′; and GAPDH: (F)5′-TCAACAGCGACACCCACTCC-3′ (R)5′-TGAGGTCCACCACCCTGTTG-3′. All primers were synthesized by Operon (Köln, Germany). All PCR reactions contained IQ™SYBR®Green Supermix from Bio-Rad, 400 nM forward and reverse primer, and 2 ng of cDNA in a total volume of 15 μL. All PCR reactions were performed in triplicate. Normalized gene expression was calculated by the treshold cycle (ΔCt) method using GAPDH as a reference.
Protein expression by western blot
Twenty-four hours after 25 nM SRFsiRNA or siSCR transfection, total protein was isolated from fibroblasts with the use of radioimmunoprecipitation assay lysis buffer from Thermo Fisher Scientific (Bonn, Germany) following manufacturer's instructions. Afterward, sodium dodecylsulfate polyacrylamide gel electrophoresis of the isolated proteins was performed and blotted on nitrocellulose membrane, which was blocked overnight in 5% bovine serum albumin. Subsequently, the membrane was incubated with a 1:500 dilution of anti-SRF and anti-actin antibody, both from Santa Cruz (Heidelberg, Germany) for 2 hours at room temperature. After washing, the second antibody from Sigma Aldrich (Seelze, Germany) with conjugated alkaline-phosphatase was applied to the membrane for 2 hours in a 1:5,000 dilution. Visualization was carried out with BCIP/NBT substrate from Sigma Aldrich. Quantification was done with ImageJ Software (National Institutes of Health), thereby the densitometry of actin and SRF were calculated and SRF was set relative to actin.
xCELLigence
The xCELLigence system from Roche (Grenzach-Wyhlen, Germany) measures electrical impedance across interdigitated microelectrodes integrated on the bottom of tissue culture E-Plates. The impedance measurement provides quantitative information about the biological status of the cells, including cell number, viability, and morphology. Therefore, 1,500–2,000 fibroblasts of 3 different patients were seeded into E-Plates™ 1 day prior transfection. Transfection was performed in quadruplicate with 25 nM of siSCR and siSRF. The impedance was measured every 30 minutes for a period of 9 days.
Proliferation assay
The 5-bromo-2′-deoxiuridine (BrdU) proliferation assay is based on the use of BrdU as a thymidine analogue. BrdU incorporates into the DNA of replicating cells and can be detected by fluorescent-labeled antibodies via flow cytometry. One day after transfection with 25 nM siSCR and siSRF, cells were incubated for 24 hours with 10 μM BrdU, and afterwards cells were detached and fixed with 70% ethanol at −20°C. Cells were denatured with 2 M hydrogen chloride and subsequently stained with phycoerythrin-labeled mouse anti-BrdU antibody (Becton Dickinson GmbH).
Cell counting
Two days after transfection with 25 nM siSCR and siSRF, the number of living cells was determined with a CASY® cell counter (Schärfe System, Reutlingen, Germany), which distinguishes dead from living cells because of their lower resistance in an electronic pulse area analysis.
Senescence assay
Senescent cells were detected by staining for senescence-associated beta galactosidase (Dimri et al., 1995). Senescence was quantified by normalization of the beta galactosidase–positive cells to the total number of living cells determined using a CASY cell counter.
Statistics
All experiments were repeated 3 times independently with different fibroblast cell lines in triplicate or quadruplicate. Statistical analysis was performed using paired t-test (2-tailed); the data (mean±standard deviation) were considered statistically significant when the P-value was <0.05 or <0.01.
Results
Isolation, cultivation and characterization of prostatic fibroblasts
The characterization of the isolated primary fibroblasts revealed a 90% expression of CD90 (Fig. 2). Additionally, fibroblasts were stained with isotype control, which did not result in a fluorescence increase (Fig. 2, left).

Characterization of CD90 expression by flow cytometry. Staining of fibroblasts with CD90 antibody reveals specific binding and fluorescence increase (right) compared to the isotype control (left). CD, cluster of differentiation.
Size and zeta potential characterization of transfection complexes
We evaluated the size and zeta potential of Interferin and siRNA transfection complexes with a 60 nM siRNA concentration. The size of the transfection complexes was 112 nm, and the zeta potential was −23.6 mV (Fig. 3A, B). These results show that the size of the transfection complexes is ideal for the transfection of cells. Additionally, the zeta potential of the transfection complexes is negative, which lets us suggest that the transfection complexes cannot mediate toxicity, which is mainly caused by highly positively charged transfection complexes, like polyethylenimine (PEI) and siRNA (Nolte et al., 2011).
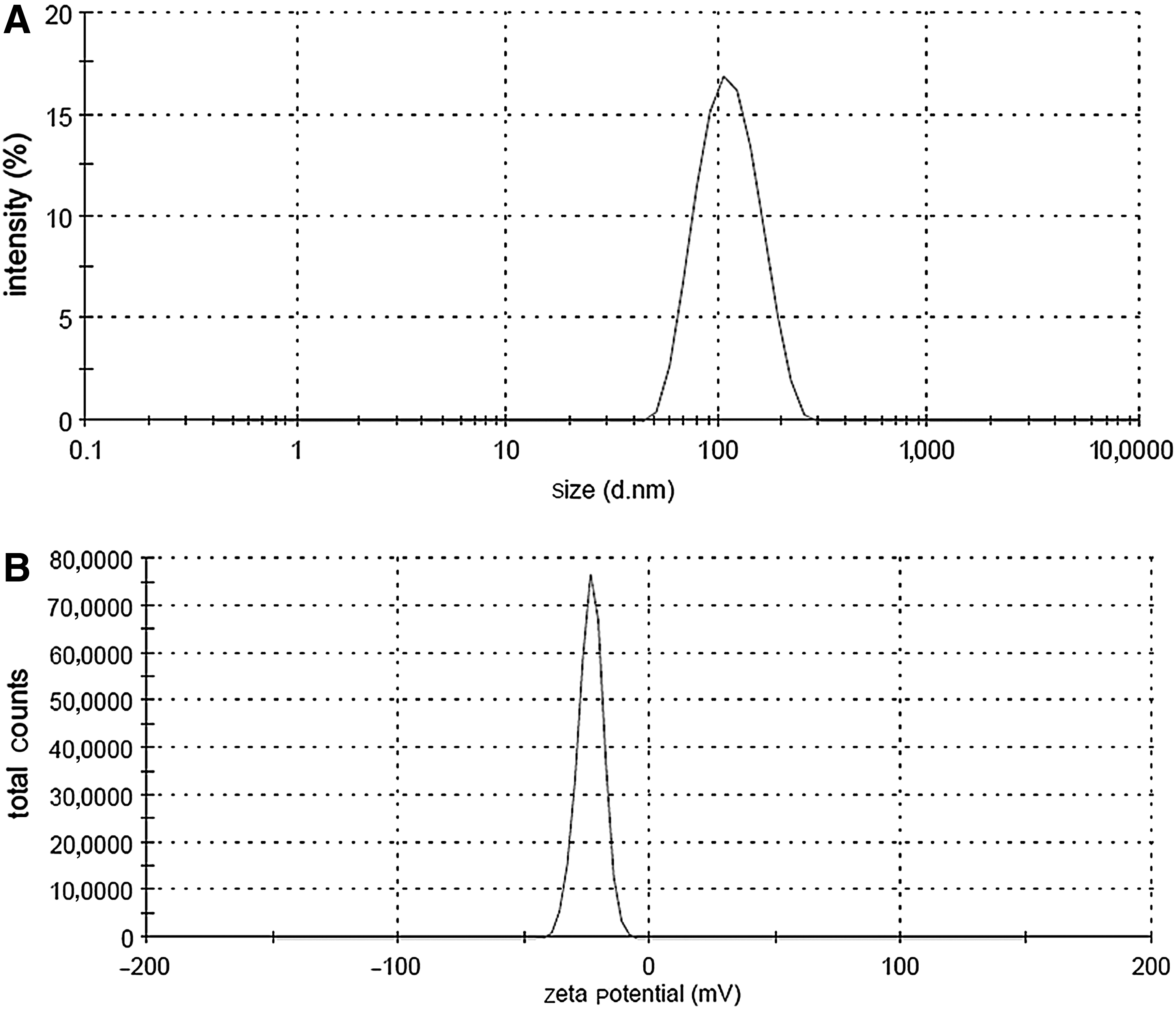
Illustration of the size and zeta potential of the transfection complexes.
SRFsiRNA-mediated silencing of SRF mRNA and protein
The transfection of fibroblasts was carried out with different siRNA concentrations of 1 nM, 10 nM, 25 nM, 50 nM, and 100 nM. The transfection with SCRsiRNA served as a control, and the expression after SCRsiRNA transfection was set to 100%. The transfection of fibroblasts with different SRFsiRNA concentrations resulted in a concentration-dependent decrease of the SRF mRNA level (Fig. 4 A, B, C). The mRNA expression decrease was specific after siSRF transfection, and the P-value for SRFsiRNA-treated cells compared with non-targeting control transfected (SCRsiRNA) cells was below 0.005 for the first transfection at all different siRNA concentrations. A saturation of the siRNA concentration seems to occur at 25 nM, because the knockdown does not increase further with increasing concentrations. Therefore we decided to use 25 nM siRNA concentrations for the subsequent cell proliferation assays.

mRNA expression of SRF after first, second, or third transfection with SCRsiRNA and SRFsiRNA.
The transfection of SRFsiRNA resulted also in a reduction of the SRF protein level. Twenty-four hours after transfection of fibroblasts with 25 nM SRFsiRNA and SCRsiRNA, we evaluated the protein level using western blot. Figure 5a shows 2 bands, a higher band that corresponds to SRF-protein, and the lower band to actin. Thereby it is visible that the SRF-corresponding band is only decreased after transfection with SRFsiRNA and not after transfection with SCRsiRNA. The knockdown of SRF was around 50% with SRFsiRNA compared with SCRsiRNA.
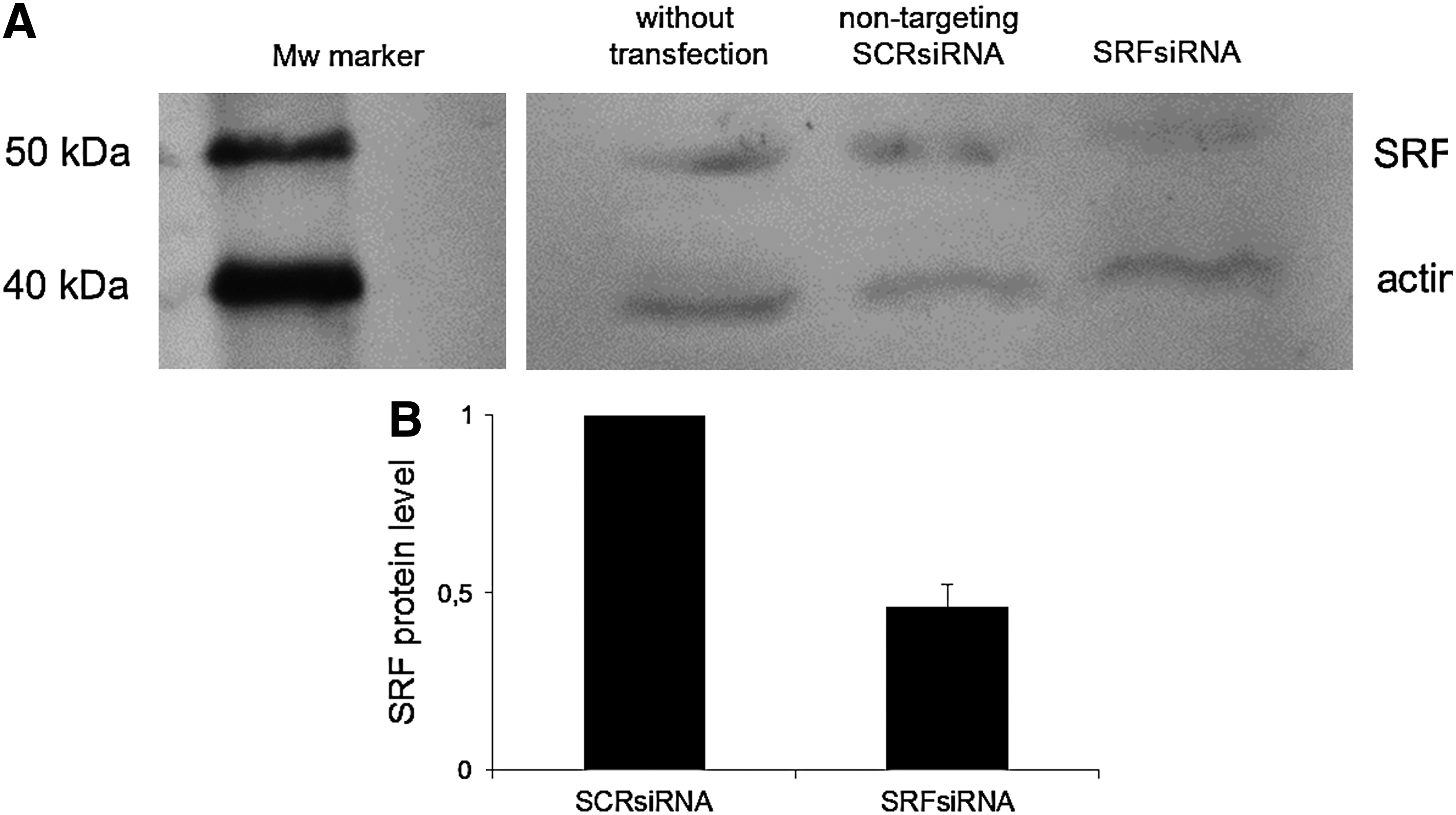
Reduction of endogenous SRF protein by SRFsiRNA one day after transfection with 25 nM siRNA concentration of SRFsiRNA and SCRsiRNA detected by western blot.
xCELLigence
Examining the fibroblasts by electrical impedance revealed increasing normalized cell index in each sample, which is the evidence for cell growth (Fig. 6). The decrease in cell index after each medium change can be explained by cell detachment from the tissue plates or shrinking cells because of insufficient warmed medium. After transfection with SRFsiRNA, fibroblasts expansion was slower, as can be seen from a smaller increase of the cell index compared with non-transfected cells. SCRsiRNA transfection leads to a higher increase of the cell index compared with non-transfected cells. Eight days after transfection, the cell index of SRFsiRNA-transfected cells was half of the cell index of SCRsiRNA-transfected cells. This suggests slower proliferation of SRFsiRNA-transfected fibroblasts compared to SCRsiRNA or non-transfected cells.
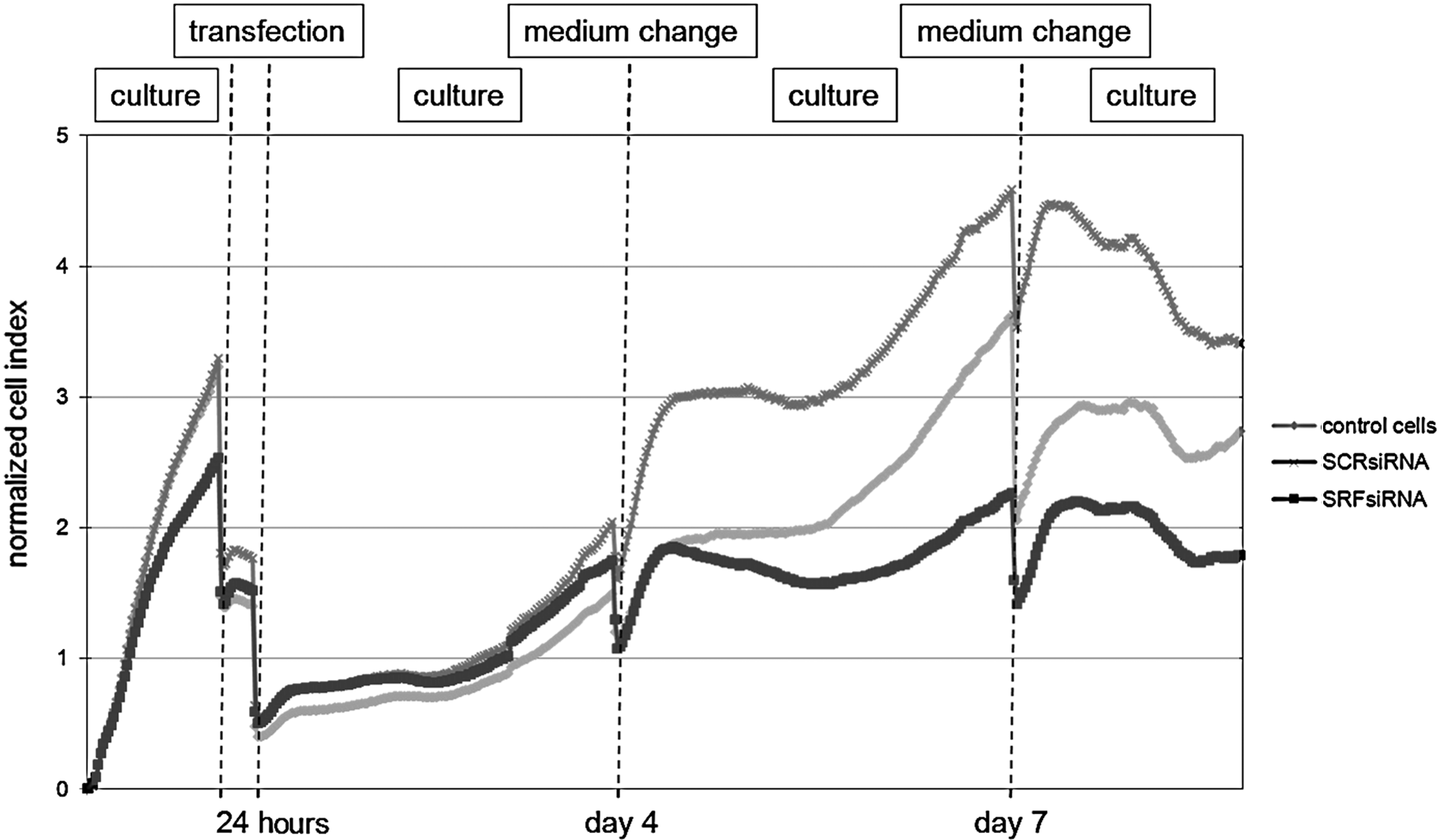
Cell proliferation measured by xCelligence. The x-axis shows the time of cell culturing, and the y-axis shows the normalized cell index, thereby increasing cell index means cell growth. Decreases occur during medium changes. Fibroblasts were either transfected with 25 nM SCRsiRNA or SRFsiRNA or not transfected to serve as a control. Fibroblast growth was examined over a timescale of 1 week after transfection.
Proliferation assay
The first SRFsiRNA transfection resulted in a 25% decrease in proliferating cells (Fig. 7A). The second and third SRFsiRNA transfections resulted in a significant decrease in cell proliferation of approximately 80%.
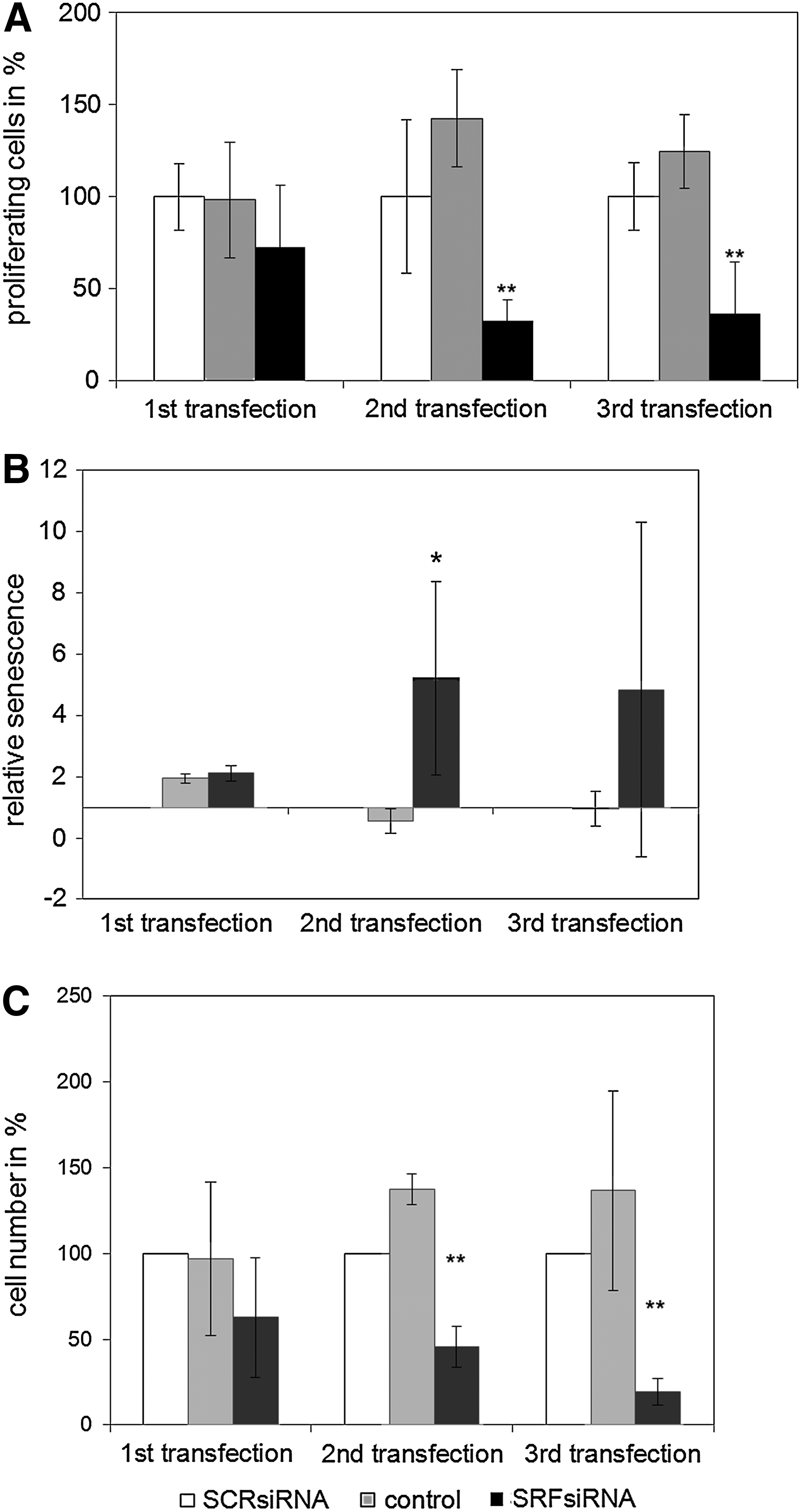
Senescence assay and cell number
After the second and third transfections of SRFsiRNA, a 5- to 8-fold increase of senescent cells was observed, whereas siSCR transfection did not increase the relative number of senescent cells (Fig. 7B).
Additionally, the cell number of SRFsiRNA-transfected cells is significantly lower compared with non-transfected cells after second (60%) and third (80%) transfections (Fig. 7C).
Discussion
SRF has been shown to control cellular processes such as migration, cell survival, and differentiation (Schratt et al., 2002; Schratt et al., 2004). Only few data exist addressing the role of SRF in primary human vascular smooth muscle cells (Chow et al., 2007; Werth et al., 2010) or in HepG2 and JHH6 hepatocellular carcinoma cells (Farra et al., 2010). This study demonstrated for the first time a correlation of SRF silencing with the inhibition of excess proliferating primary prostatic fibroblasts.
The characterization of the cells revealed a pure fibroblast cell population positive for CD90 expression (Fig. 2).
The transfection complexes have a size of 112 nm, which is favorable for the transfection of cells. Additionally, the zeta-potential of 60 nM siRNA transfection complexes is negatively charged at −23.6 mV. The negative charge of the transfection complexes lets us conclude that the positive charge of the Interferin is neutralized at already low siRNA concentrations. Furthermore, the mediation of cytotoxicity from the transfection reagent can be excluded by that result. In comparison, the transfection of siRNA and PEI can cause high toxicity if transfection complexes have a highly positively charged surface (Boussif et al., 1995).
The presented data indicate that SRFsiRNA is able to reduce SRF expression in primary fibroblasts, confirmed by the significant decrease in SRF mRNA (Fig. 4A, B, C). Recently, Werth et al. transfected human primary vascular smooth muscle cells with the same SRFsiRNA leading to decreased mRNA and protein levels of SRF (Werth et al., 2010). Our results show further that the knockdown depends on the siRNA concentration. One-time transfection with a concentration of 10 nM siRNA results already in a knockdown of 67% compared with non-targeting control (SCRsiRNA) (Fig. 4A). From previous studies in our lab concerning the transfection reagent, we were also able to show a saturation of the siRNA at a concentration of 25 nM (Nolte et al., 2009). Besides, we decided to use the lowest efficient concentration of 25 nM to minimize cytotoxic effects of the transfection reagent that can occur at 100 and 150 nM siRNA concentrations (Nolte et al., 2009). Additionally, the transfection of 25 nM siRNA concentrations is low compared with other groups that use different cationic polymers like PEI (Nolte et al., 2011) (250 mM), Chitosan (Chen et al., 2012) (64 nM), or liposomal transfection (Rothdiener et al., 2010) (600 nM to 2.5 μM).
The transfection of siSCR does not mediate a knockdown of the SRF mRNA level, but rather, a significant up-regulation only after the first transfection (Fig. 4A).
A detailed look at the results shows also an up-regulation caused by the transfection of SCRsiRNA in the Xcelligence data. The evaluation of the cell number, cell proliferation, and senescence show contrary data. In detail, after 1 transfection with SCRsiRNA the xCelligence shows improved cell proliferation, but this influence is visible for the first time after 3 days. In the experimental setup of the cell number, cell proliferation, and senescence, the cells were already transfected after 3 days for the second time. Herein, the results show no up-regulation of the cell number, cell proliferation, or senescence, compared with non-transfected cells. In contrast, cells transfected with SCRsiRNA show decreased cell number, cell proliferation, and senescence after the second and third transfections. Based on this result we assume a sequence independent effect, like a small stress response to the transfection procedure itself.
To our knowledge, the transfection reagent alone may not be responsible for the up-regulation of SRF; previously, we tested Interferin without siRNAs at different concentrations and found no up-regulation of the E-selectin expression in human primary endothelial cells (Nolte et al., 2009). We assume that SRF up-regulation after the first transfection is only noticeable by mRNA level measurement after the first transfection and xCelligence and is caused by a combined effect of the SCRsiRNA and Interferin, which could not be noticed in previous studies (Nolte et al., 2009; Nolte et al., 2011).
The transfection of SRFsiRNA also resulted in an SRF protein level decrease of around 50%, as shown by western blot analysis (Fig. 5). Recently, Farra et al. were able to show a significant decrease of the SRF protein level in hepatocellular carcinoma cells HepG2 and JHH6 with the same siRNA sequence targeted against SRF (Farra et al., 2010). We are aware that off-target effects triggered by control siRNAs, targeted siRNAs, and transfection reagents cannot be entirely ruled out and previous work suggests examining further siRNAs targeted against SRF, SCRsiRNA controls and different transfection reagents to support our results.
The down-regulation of SRF leads to permanent slower cell proliferation and DNA replication examined by xCELLigence (Fig. 6) and BrdU assay (Fig. 7A) respectively. The transfection of SRFsiRNA does also lead to a significant increase of senescent cells after the second transfection (Fig. 7B). Additionally, the total cell number of SRFsiRNA-transfected cells is much lower after the second and third transfections compared with control cells (Fig. 7C).
Our results support the work of Werth et al. and Farra et al., who showed a significant reduction in cell proliferation in human smooth muscle cells or hepatocellular carcinoma cells after SRFsiRNA transfection (Farra et al., 2010; Werth et al., 2010), leading to the conclusion that SRF expression has a significant impact on cell proliferation. Taken together, our results indicate that SRF silencing results in a permanent slowdown of fibroblast proliferation, which could be useful for clinical applications. The need for several transfections to achieve a significant biological effect is no disadvantage for therapeutic application. We aim to immobilize siRNA into a biomaterial on a catheter to enable a local release of siRNAs from the catheter to the site of action for several days or weeks. For this therapeutic application, the need for more than 1 transfection would be no disadvantage. Furthermore, we assume that a significant siRNA silencing efficiency in vitro and in vivo is around 2–3 days. For this reason, a longer release of siRNAs might be favorable in any event. Additionally, we could prove by xCelligence that the cell proliferation is already decreased within 3 days after 1 transfection with SRFsiRNA.
Previous studies for local in vivo siRNA applications focus on diminishing immunoreactivity of implants with a local administration of siRNA (Nolte et al., 2011).
Herein, the poor long-term outcome of urethrotomy might be improved by the immobilization of SRFsiRNA on a catheter. Further studies will clarify this possible new approach to combat restenosis of urethral strictures after urethrotomy.
Conclusion
We were able to prove that SRF knockdown leads to permanent slowdown of cell proliferation in excess proliferating prostatic fibroblasts in vitro. This impact on SRF silencing opens the way to locally treat urethral strictures with siRNA and lead to reduced fibroblast proliferation, resulting in prolonged patency of the urethra. Our findings confirm RNAi therapy as a promising new strategy to improve the long-term outcome after urethrotomy. As next animal experiments have to confirm the positive effects of SRFsiRNA to prevent from restenosis of the urethra.
Footnotes
Acknowledgments
We thank Roche (Grenzach-Wyhlen, Germany) for kindly providing their xCELLigence System for cell proliferation measurement. We also like to thank Sandra Stoppelkamp for her excellent proof reading of the manuscript. Additionally, we have to thank Jörg Hennenlotter for providing the native prostatic material. We would also like to thank Besmire Sutaj for conducting the western blot analysis.
Author Disclosure Statement
No competing financial interests exist.