
Abstract
Small interfering RNAs (siRNAs) can trigger potent gene silencing through the RNA interference (RNAi) pathway. The RNA-induced silencing complex (RISC) is key to this targeted mRNA degradation, and the human Argonaute2 (hAGO2) endonuclease component of RISC is responsible for the actual mRNA cleavage event. During RNAi, hAGO2 becomes loaded with the siRNA guide strand, making several key nucleic acid-enzyme interactions. Chemically modified siRNAs are now widely used in place of natural double-stranded RNAs, and understanding the effects chemical modifications have on guide strand-hAGO2 interactions has become particularly important. Here, interactions between the 5′ nucleotide binding domain of hAGO2, MID, and chemically modified nucleotide analogues are investigated. Measured dissociation constants reveal that hAGO2 does not discriminate between nucleotide analogues during binding, regardless of the preferred sugar conformation of the nucleotide analogues. These results correlate well with cell-based gene silencing results employing siRNAs with 5′-modified guide strands. Additionally, chemical modification with 2′-deoxy-2′-fluoroarabino nucleic acid (2′F-ANA) and 2′-deoxy-2′-fluororibonucleic acid (2′F-RNA) at the passenger strand cleavage site of siRNAs has been shown to prevent hAGO2-mediated strand cleavage, an observation that appears to have little impact on overall gene silencing potency.
Introduction
Elucidating the interactions between the siRNA guide strand and hAGO2 has been an important area of study in recent years, especially in light of siRNA-based drug development programs demonstrating successful late-stage clinical trials. The solved structures of AGO enzymes with bound guide strands and RNA targets have been reported (Wang et al., 2008a; Wang et al., 2008b; Wang et al., 2009), including a recent reports describing human Argonaute2 (Schirle and MacRae, 2012; Elkayam et al., 2012). hAGO2 has 4 domains: N-terminal, PAZ, middle (MID), and PIWI. PIWI contains a ribonuclease (RNase) H-like fold, and performs the mRNA cleavage event. The PAZ domain interacts with the 3′ end of the loaded guide strand, and the MID domain interacts with the 5′ end. Interactions between the guide strand 5′ end and the MID domain have been studied (Frank et al., 2010), revealing preferential binding of uracil (U) and adenine (A) 5′ nucleotides over guanine (G) and cytosine (C). Nuclear magnetic resonance (NMR) titration and x-ray crystallographic studies have identified a 30-fold higher affinity for binding of A and U versus G and C in hAGO2, and demonstrated clear discriminating interactions between the nucleobases of nucleotide monophosphates and a rigid loop in the MID domain.
siRNA-based drug platforms face 3 significant challenges: (1) nucleic acids have poor nuclease stability, (2) siRNAs are not easily delivered to target cells, and (3) siRNAs can cause off-target effects and immunostimulation. Chemical modification of the siRNA backbone has become increasingly common, and an impressive library of chemically modified nucleotide analogues has been developed for improving the nuclease stability and off-target effects/immunostimulation characteristics of siRNAs (Deleavey and Damha, 2012). To advance these efforts and to investigate the effects of guide strand chemical modifications on the interactions between hAGO2 and the siRNA guide, we have employed a dinucleotide-based model to examine relative binding affinities of RNA, 2′-deoxy-2′-fluoroarabino nucleic acid (2′F-ANA), 2′-deoxy-2′-fluororibonucleic acid (2′F-RNA), and DNA nucleotides for the MID domain of hAGO2. Results demonstrate that the MID domain tolerates chemically modified nucleotide analogues, regardless of their preferred sugar conformation. As well, switching uracil with thymine nucleobases has little effect on binding affinity. Measured affinities are in agreement with cell-based gene silencing results employing chemically modified siRNAs.
Previously, siRNAs modified with 2′F-ANA and 2′F-RNA have proven particularly useful (Dowler et al., 2006; Deleavey et al., 2010). In siRNA, 2′F-ANA enhances siRNA nuclease stability, can be readily incorporated in the siRNA passenger strand, and can be used in combination with the rigid North-type nucleoside analogues 2′F-RNA and locked nucleic acid to create fully modified siRNAs with improved potency, reduced immunostimulatory properties, and a thermodynamic bias for antisense strand RISC loading (Dowler et al., 2006; Deleavey et al., 2010). The binding studies presented here demonstrate that both 2′F-ANA and 2′F-RNA are tolerated at the guide strand 5′ end (the MID binding site), as well as DNA. Additionally, 2′F-ANA/2′F-RNA at the siRNA passenger strand cleavage site prevents hAGO2-mediated strand cleavage but appears to have little impact on overall gene silencing potency.
Materials and Methods
Oligonucleotide dimer synthesis
Dimer nucleotides (5′ phosphorylated, 3′-OH) were synthesized on an Applied Biosystems (ABI) 3400 DNA synthesizer at 1μmol scale. Unylink CPG (ChemGenes) was used for the syntheses of all chemically modified dimers (synthesis of RNA dimers can be found in the supporting information). 2′F-ANA and 2′F-RNA phosphoramidites were prepared as 0.15 M solutions in dry acetonitrile (ACN), and DNA phosphoramidites were prepared at 0.1 M. For addition of the 5′ phosphate, bis-cyanoethyl-N,N-diisopropyl phosphoramidite (ChemGenes) was used at 0.2 M. 5-ethyltetrazole (0.25 M in ACN, ChemGenes) was used to activate the phosphoramidites for coupling. Detritylations were accomplished with 3% trichloroacetic acid in dichloromethane for 110 seconds. Capping of failure sequences was achieved with acetic anhydride in tetrahydrofuran, and 16% N-methylimidazole in tetrahydrofuran. Oxidations were done using 0.1 M I2 in 1:2:10 pyridine:water:tetrahydrofuran. Phosphoramidite coupling times were 600 seconds for 2′F-ANA T and 2′F-RNA U and 900 seconds for 2′F-ANA and 2′F-RNA G. DNA coupling times were 110 seconds for T and 270 seconds for G. The 5′ phosphorylation reagent was coupled for 1200 seconds.
Twenty-one nucleotide oligonucleotides used for firefly luciferase gene silencing and hAGO2 strand cleavage assays were prepared according to standard protocols, as described in Deleavey et al. (2010).
Oligonucleotide dimer cleavage and deprotection
Base deprotection and cleavage from the solid support was accomplished with 1 mL of 3:1 aqueous NH4OH:EtOH for 48 hours at room temperature, after which samples were vented for 2 hours, chilled on dry ice for 15minutes, and lyophilized to dryness in a speedvac concentrator (Savant).
Oligonucleotide dimer purification and characterization
All dimers were purified by reverse phase high-performance liquid chromatography (HPLC) on an Agilent 1200 series HPLC system using a Waters μBondapak semipreparative C18 column. A stationary phase of 100mM triethylammonium acetate in water with 1% ACN (pH 7), and a mobile phase of HPLC-grade ACN (Sigma) were used (with a gradient of 1%–15% over 15 minutes). Purified oligonucleotides were lyophilized to dryness, resuspended, and lyophilized again, which served to remove excess triethylammonium acetate salts. Dimers were characterized by electrospray ionization–time-of-flight mass spectrometry (results provided in Supplementary information Table S1; Supplementary Data are available online at www.liebertpub.com/nat) carried out on a QTOF22 (Micromass) from Waters in negative mode.
MID domain preparation
The MID domain of hAGO2 (residues 439 to 578) was prepared as described previously (Frank et al., 2010). Briefly, MID domain was expressed in Escherichia coli with an N-terminal His6-Sumo-tag (Mossessova and Lima, 2000) and purified via nickel-nitroloacetic acid affinity chromatography. The tag was then cleaved using Ulp-1 protease and the protein was further purified via cation exchange and size exclusion chromatography. Buffer composition for storage and experiments was 25 mM 2-(N-morpholino) ethanesulfonic acid pH 6.5, 200 mM NaCl, and 1 mM dithiothreitol (DTT).
Human AGO2 preparation
Full-length hAGO2 (starting at residue 34) was prepared as previously described (Frank et al., 2011). Briefly, hAGO2 was expressed using the FastBac system (Invitrogen) for protein production in SF9 insect cells. Baculovirus-infected cells were harvested 72 hours post-infection and protein was purified via NiNTA-affinity chromatography. This was followed by cleavage of the tag using tobacco etch virus protease and Superdex-200 size-exclusion chromatography in 25 mM Tris pH 8.0, 300 mM NaCl, 3 mM DTT, and 5 % glycerol.
NMR titrations
NMR titration experiments were carried out at 293 K using a Bruker 600 MHz spectrometer. NMR titrations were performed by acquiring 1H-15N heteronuclear single quantum correlation (HSQC) spectra on samples of ∼0.10 mM 15N-labeled hAGO2 MID domain with addition of increasing amounts of dinucleotide ligand. Significantly shifting peaks were selected, and their coordinates in the spectra at different ligand concentrations determined. Chemical shift differences were calculated as Δδ=[(ΔδH)2+(0.2×ΔδN)2]1/2, where ΔδH and ΔδN are the observed chemical shift changes for 1H and 15N, respectively. The chemical shift difference (i.e. the distance travelled by a peak after addition of ligand, is proportional to the amount of bound ligand. Therefore, for determination of dissociation constants, Δδ was plotted as a function of the molar ratio (nucleotide/protein), and the data was fit using the maximum shift and dissociation constant as adjustable parameters. Each titration experiment was performed once, and 3 significantly shifting peaks were identified in each experiment. Peaks used for the determination of dissociation constants for each dimer are provided (See Fig. 1 and Supplementary Information), marked with arrows. For each dimer, the fits from 3 unique shifting signals were all used to calculate mean solution dissociation constant, shown with associated standard deviations (bars).

Binding affinities of natural and chemically modified dinucleotides for the 5′ binding of the middle (MID) domain.
Firefly luciferase gene silencing assays
Firefly luciferase assays were carried out in HeLa X1/5 cells stably expressing firefly luciferase. siRNA duplexes targeted firefly luciferase mRNA from position +1818 to +1836, and were delivered using Lipofectamine-Plus (Invitrogen). These assays were conducted according to previously described studies (Deleavey et al., 2010) and are shown in Fig. 2.

Gene silencing effects of small interfering RNAs (siRNAs) with chemically modified guide strand 5′ termini.
Target strand cleavage assays
Human AGO2 target cleavage assays were carried out using 5′-radiolabeled 21-nt target RNA or fluorinated target (see Fig. 3a). Target RNA was labeled using T4 polynucleotide kinase and [γ- 32P]ATP. Radiolabeled RNA was then purified by phenol/chloroform extraction and ethanol precipitation. For slicing assays, first, 100nM full-length hAGO2 was incubated at 37
Results and Discussion
Several previously reported crystal structures have shown that the 5′ phosphate of the guide strand is tightly bound to the MID domain, bending the 5′ guide strand nucleotide out and away from the rest of the guide strand, making it unavailable for the target mRNA base-pairing interactions seen in the neighboring seed region nucleotides (Wang et al., Wang et al., 2008a; Wang et al., 2009; Frank et al., 2010). These crystal structures also suggest an interesting structural preference for 5′ ribonucleotides bound in the MID domain: the guide strand 5′ ribonucleotide appears to adopt a DNA-like south (C2′ endo) sugar conformation in this binding pocket (Wang et al., 2008a; Wang et al., 2009; Frank et al., 2010; Schirle and MacRae, 2012). It is well known that 2′F-ANA has a preference to adopt south and eastern sugar conformations, and 2′F-RNA prefers a north-type sugar pucker. Both of these chemical modifications feature 2′-fluorine substitution, which introduces unique electron-withdrawing properties in duplexes (Manoharan et al., 2011), changes in oligonucleotide duplex hydration (Watts and Damha, 2008; Manoharan et al., 2011), and removes the hydrogen bond–donating 2′-OH functionality of RNA. The 2′F-ANAs and DNAs used in this study featured thymine nucleobases, while 2′F-RNAs and RNAs carried the uracil nucleobase. All of these differences could potentially impact MID-domain binding affinity and, by extension, gene silencing potency. Indeed, hAGO2-guide strand binding affinity and target cleavage activity can be affected by interactions between hAGO2 and the first two 5′ nucleotides of the RNA guide strand (Lima et al., 2009).
A series of 5′ phosphorylated dimers, all 5′-pUpG-3′ or 5′-pTpG-3′ in sequence, were designed and synthesized to be tested in a model hAGO2 binding system employing purified MID domain (Fig. 1). This system has previously been reported for binding of 5′ ribonucleotide monophosphates (Frank et al., 2010). These dimers match the predicted best binding sequence from bioinformatics and previous NMR studies (Frank et al., 2010), and employing dimers in place of mononucleotides was predicted to improve binding. NMR titration experiments were used to measure the dissociation constants (KD) for the binding of each dimer to the MID domain. Results are shown in Fig. 1, which demonstrate that 5′ phosphorylated, 3′-OH dimers bind the MID domain approximately 2.5-fold more tightly than 5′-ribonucleotide monophosphates reported previously (∼120 μM versus ∼50 μM KD) (Frank et al., 2010). This increase in affinity is explained by the additional contacts the second nucleotide makes within the MID domain as were observed in the structures of full-length hAGO2 in complex with guide RNA (Elkayam et al., 2012; Schirle and MacRae, 2012). Nitrogen-15 HSQC NMR spectra for each dimer titration experiment (along with plots used for determining dissociation constants) are provided in the Supplementary Information; 15N HSQC NMR spectra for 5′-pUpG-3′-OH are shown in Fig. 1. Titration of dimers introduces the same peak shifts as seen previously for nucleoside monophosphates; however, additional shifts are obtained from the additional contacts that the second nucleotide provides in the MID domain.
Interestingly, the MID domain shows no strong preference for binding with any of the sugar modifications tested; RNA, DNA, 2′F-ANA, and 2′F-RNA all bind with comparable dissociation constants. DNA and 2′F-ANA dimers feature thymine nucleobases, whereas RNA and 2′F-RNA carry uracil; this substitution also does not appear to have an effect on dissociation constants, demonstrating that the MID domain of hAGO2 is tolerant to chemically modified nucleotide analogues that do not feature bulky substitutions, regardless of preferred sugar conformation.
To determine if these observed similarities in MID domain binding affinity translate from this model system to actual gene silencing potencies in cell culture, firefly luciferase-targeting siRNAs (previously studied in detail in Deleavey et al., 2010) were chemically modified at the guide strand 5′ termini to match several of the dimers tested in Fig. 1. Each of the siRNAs tested, and their gene silencing knockdown effects, are presented in Fig. 2. Because this study was designed to investigate the interactions between the RISC complex and chemically modified siRNAs, a previously identified potent chemically modified siRNA design reported in (Deleavey et al., 2010) was also included for reference (Luc 2′F-ANA/2′F-RNA). These gene-silencing assays clearly demonstrate that guide strand 5′ modification with DNA, 2′F-ANA, or 2′F-RNA has little impact on gene silencing potency, especially at the higher siRNA concentrations tested (50 nM to 0.4 nM). These findings are in line with the dimer-MID binding affinity findings. It has been previously observed that full guide strand modification with 2′F-ANA is detrimental for gene silencing potency (Deleavey et al., 2010), and these studies demonstrate that reductions in potency are unlikely to originate from impaired interactions at the guide strand 5′ terminus. Reduced tolerance for 2′F-ANA guide strands more likely originates elsewhere in hAGO2, perhaps near the mRNA cleavage site of the PIWI domain. 2′F-ANA is likely well tolerated in the 3′-binding PAZ domain (Dowler et al., 2006).
The fate of the siRNA passenger strand during RISC loading with chemically modified siRNAs is not well defined. During RISC loading with unmodified siRNA, the passenger strand is typically cleaved by the PIWI subdomain of hAGO2, followed by unwinding from the guide strand. There are possible bypass mechanisms as well, in which the intact passenger strand can be unwound by ATP-dependent helicase activity (Gaynor et al., 2010). It is well known that the PIWI subdomain of hAGO2 contains an RNase H-like fold, which has endonuclease activity dependent on 2 magnesium ions and catalyzes passenger strand (and subsequently, mRNA target) cleavage (Liu et al., 2004; Gaynor et al., 2010; Deleavey and Damha, 2012). hAGO2-mediated oligonucleotide cleavage produces a 3′-OH fragment and a 5′-phosphate fragment (Martinez and Tuschl, 2004), suggesting that a 2′ hydroxyl group available to form the 2′–3′ cyclic phosphate observed for some other nucleases is not required. In some cases, but not always, interfering with passenger strand cleavage through 2′ modifications at the scissile phosphate can reduce gene silencing potency (Martinez and Tuschl, 2004; Matranga et al., 2005; Muhonen et al., 2007).
To examine the process of hAGO2 loading with a 2′F-ANA/2′F-RNA–modified siRNA (corresponding to a potent siRNA design reported in Deleavey et al., 2010), the ability of 2′F-RNA guide strand–loaded hAGO2 to cleave a 2′F-ANA/2′F-RNA–modified target strand was investigated. The experimental design, shown in Fig. 3, consisted of 2 steps: first, purified hAGO2 was loaded with a guide strand (introduced as a single strand, either a 2′F-RNA–modified sequence or a control let-7 RNA), and then a complementary target was introduced (either an RNA complement, or a 2′F-ANA/2′F-RNA–modified target strand). The 2′F-ANA/2′F-RNA target strand corresponds to the chemical modifications of the passenger strand of the Luc 2′F-ANA/2′F-RNA siRNA from Fig. 2. Results demonstrate that 2′F-RNA-guided hAGO2 is able to readily cleave a complementary RNA target, whereas a 2′F-ANA/2′F-RNA–modified target strand is not easily cleaved. These results suggest that hAGO2 cleavage activity is impaired for substrates modified with 2′F-ANA and 2′F-RNA on opposite sides of the scissile phosphate. hAGO2 cleavage occurs opposite nucleotides 10 and 11 of the guide strand measured from the 5′ end (Soutschek et al., 2004; Kraynack and Baker, 2006; Ui-Tei et al., 2008; Judge et al., 2009), and so the target cut site is between the
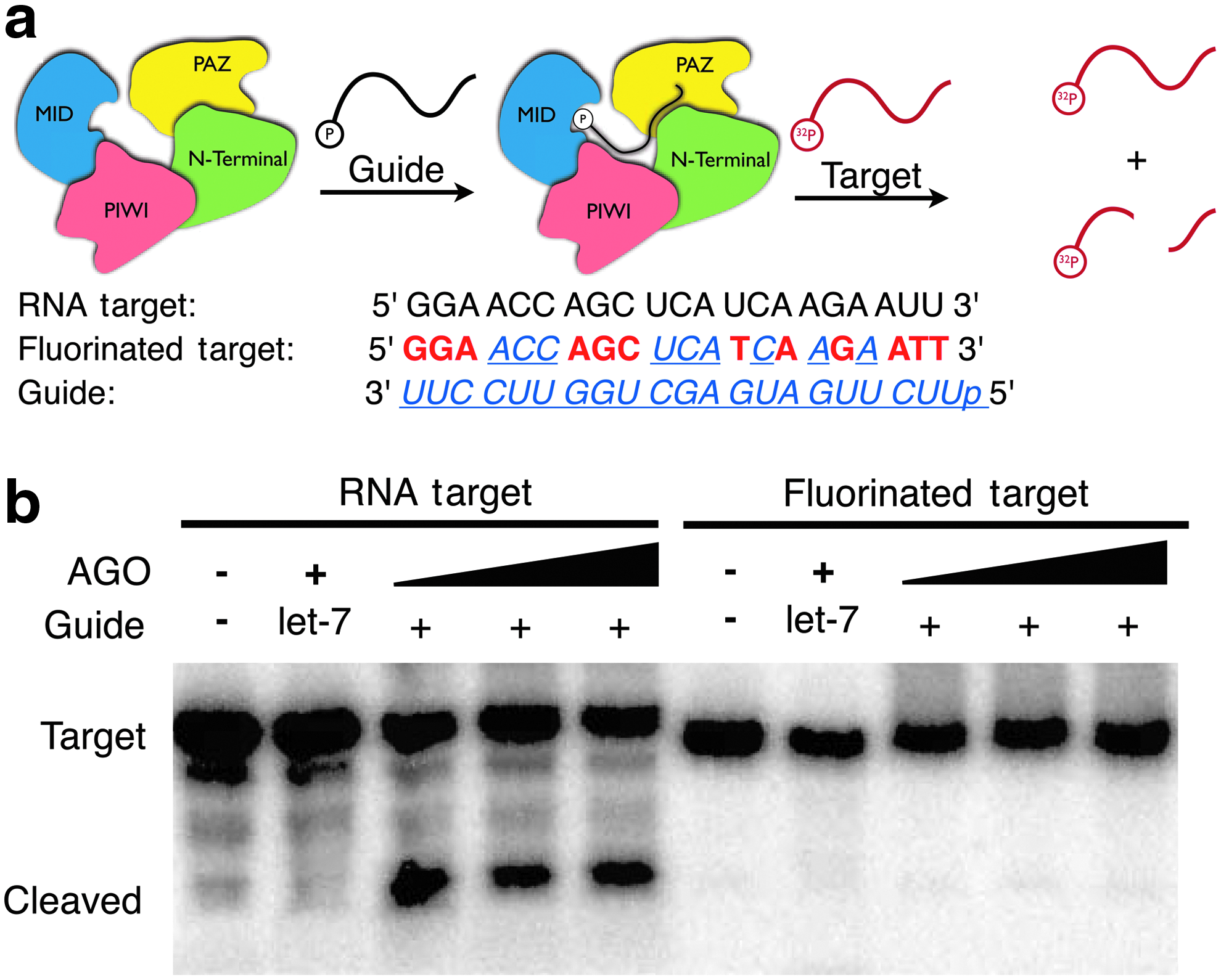
Human Argonaute2 (hAGO2)-mediated cleavage of RNA and 2′F-ANA/2′F-RNA-modified targets.
Conclusions
Chemically modified siRNAs are becoming increasingly important to several siRNA-based drug discovery programs. As a result, additional insights into the interactions between the RISC complex and chemically modified siRNAs are necessary. Additionally, understanding the details surrounding the passenger strand's fate during RISC loading with chemically modified siRNAs might assist in the drug development process. Here, binding studies between the 5′ nucleotide binding domain of hAGO2, MID, and chemically modified nucleotide analogues have been reported. Dissociation constants reveal that hAGO2 does not discriminate between nucleotide analogues during binding, regardless of the preferred sugar conformation of the nucleotide analogues. These results correlate well with cell-based gene silencing results employing siRNAs with 5′-modified guide strands. Additionally, chemical modification with 2′F-ANA and 2′F-RNA at the passenger strand cleavage site of siRNAs has been shown to prevent hAGO2-mediated strand cleavage, an observation that appears to have little impact on overall gene silencing potency. Together, these findings shed some light on the use of chemically modified nucleotide analogues in siRNA modification and contribute to the rapidly advancing field of oligonucleotide-based gene silencing therapeutics.
Footnotes
Acknowledgments
Masad J. Damha and Bhushan Nagar were supported by an operating grant from the Canadian Institutes for Health Research. Glen F. Deleavey was supported by a Vanier Canada Graduate Scholarship. Some NMR experiments were recorded at the Québec/Eastern Canada High Field NMR Facility, supported by the Natural Sciences and Engineering Research Council of Canada, the Canada Foundation for Innovation, the Québec ministère de la recherche en science et technologie, and McGill University. We would like to thank Altus Formulation for conducting the firefly luciferase gene silencing assays.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.