
Abstract
Molecular barcode arrays are widely employed in the analysis of large strain libraries, whereby probes linked to unique oligonucleotides (“antitags”) are used to detect selected DNA targets (“tags”) by highly specific hybridization. One of the major problems for such screen designs is thus insuring a high degree of probe–target specificity and a low level of nonspecific binding (in sum, “orthogonality”) across the entire tag population (“collection”). Several approaches have been previously proposed for designing orthogonal DNA tags by–among others–focusing on their individual or pair-wise structures, such as Smith Waterman sequence similarity, the widely used nearest neighbor method, and full thermodynamic estimates of sequences. However, these methods generally involve imposing various heuristic constraints (“design rules”) on possible tag/antitag (TaT) sequences in order to achieve probe–target specificity across the collection. The resulting lack of freedom in considering all putative sequences can lead to potentially suboptimal designs and to the ensuing reduction in the degree of orthogonality within the constructed TaT collections. Here, we demonstrate that a randomized-search algorithm based on simulated annealing optimization can be used in order to substantially free the design process from the limitations of sequence constraints—allowing for the elucidation of potentially more optimal DNA tag collections. The designed sets of DNA oligonucleotides are optimized for the highest degree of orthogonality as quantified by melting temperature Tm—an experimentally relevant system property, which could also be used as a theoretically meaningful thermodynamic metric for optimizing TaT binding specificity. That is, this work describes an approach to constructing tag/antitag libraries, which offer the greatest melting temperature separation between specific probe–target duplexes and other nonspecific structures. The proposed method finds, with high probability, the global solution that maximizes the difference in Tm between the specific and nonspecific tag–antitag hybridizations across a collection of given size for TaTs of specified length. An application of this approach is demonstrated using 2 different DNA probe sets.
Introduction
For instance, in DNA microarray applications, a set of oligonucleotides is immobilized at specific locations on chip surface to serve as probes for the presence of specific nucleotide sequences. The solution containing tagged target samples is then brought in contact with microarray surface to hybridize with probe antitags. The unbound molecules are subsequently washed out, while those that remain hybridized are monitored by scanning the chip. Since each location on the chip corresponds to a specific probe oligonucleotide, expression data is elucidated by measuring signal intensities of sample-specific labels at each location.
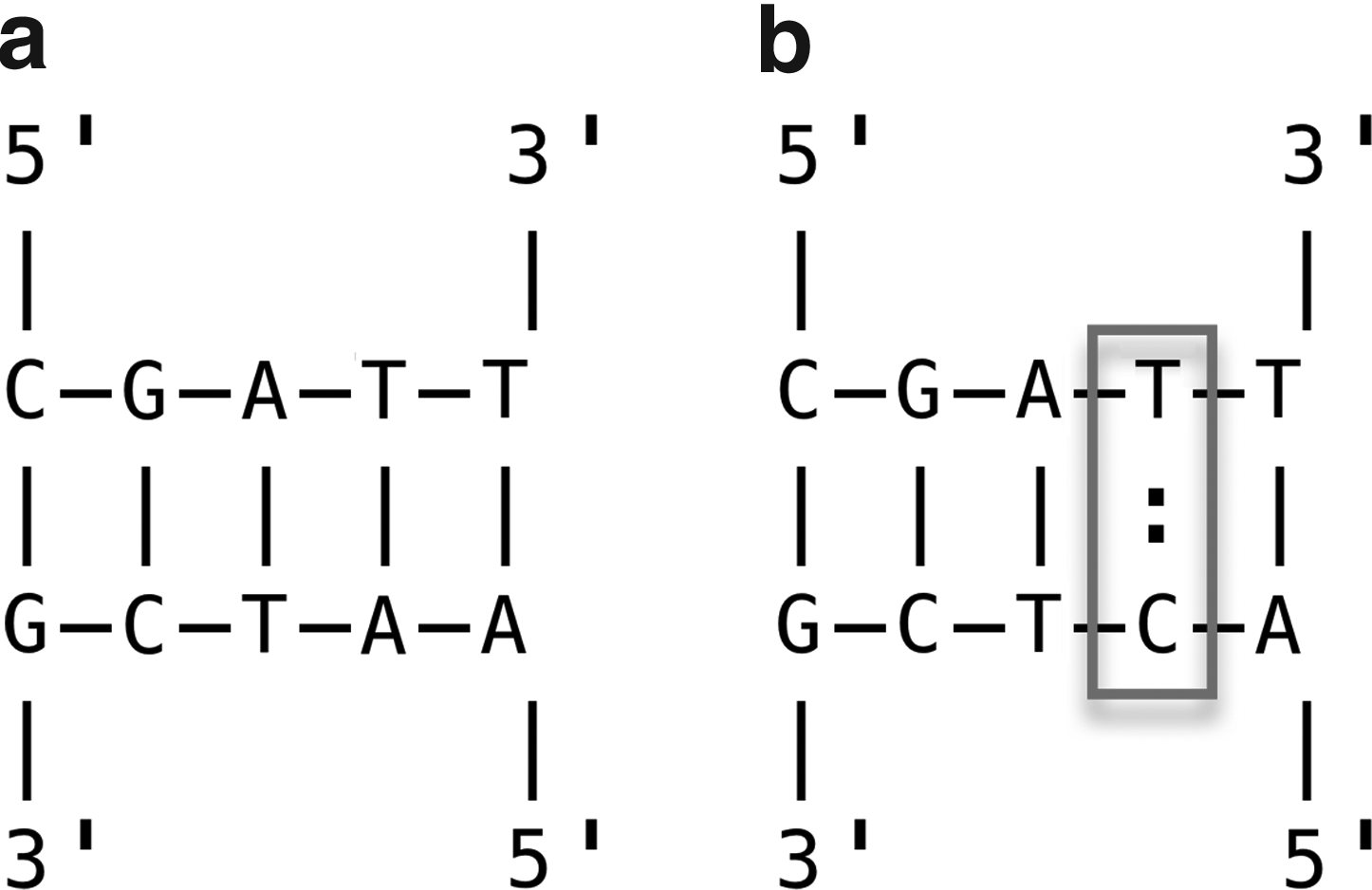
In this context, we call an event when a probe hybridizes to its own (Watson-Crick complementary) target a “perfect-hybridization”. When such perfect-hybridization reactions predominate, an assay is likely to produce a strong signal specific to target's label that results in its accurate identification (Fig. 1a). However, when probes spuriously hybridize to wrong targets or to each other, “cross-hybridization” events occur—leading to misidentification of the target by the noncognate probe or to signal reduction due to probe–probe sequestration (Fig. 1b). (Analogously, we also refer to a pair of cross-hybridized sequences as “cross-hybrid” and perfect-hybridized sequences as “perfect-hybrid” in subsequent discussion.) Therefore, a reliable identification assay requires a tag/antitag library (“collection”) that is optimized to exhibit strong perfect-hybridization and weak cross-hybridization for any possible pairing among its individual elements (i.e., it has to be orthogonal) (Ben-Dor et al., 2000).
In this work, we develop a method for finding a set of DNA oligonucleotides with the highest degree of orthogonality—for a specified set size/sequence length and without the need to impose any heuristic design constraints on oligonucleotide composition—by using an algorithm based on randomized-search simulated annealing. Note that the definition of “orthogonality” is subject to the specification of the metric with which the amount of perfect- versus cross-hybridization is quantified. Here, we employ melting temperature (Tm) as the thermodynamic basis for such a metric. While previous works have often used other system parameters, such as the free energy of hybridization (ΔG) or reaction equilibrium constants, we propose to target melting temperature for optimization since it could be most readily controlled in experimental settings, thus offering significant advantages in practical applications.
System model for orthogonal tags
Oligonucleotide orthogonalization
Let us begin by considering 2 DNA oligonucleotides: antitagU and antitagV (with their Watson-Crick complements referred to as tagU and tagV, respectively). This pair is taken to be “orthogonal” when the highest measure of its cross-hybrid affinities is much less than the lowest affinity of its perfect-hybrids (i.e., the highest affinity among antitagU–antitagV, tagU–tagV, antitagV–tagU, and antitagU–tagV dimers has to be much lower than the lowest affinity of either antitagU–tagU or antitagV–tagV duplex). This will lead dimerization reaction conditions to favor the production of desirable perfect-hybrid duplexes over the undesirable cross-hybrid ones. (In our application, melting temperature will be used as a measure of affinity, as discussed below.) While this may be easily achieved for the case of 2 antitags, the problem becomes significantly more difficult as the desired size of the library to be designed (i.e., the number of simultaneously resolvable elements) increases. The 2 problems involved are the presence of (1) near-complementary antitags, and (2) sequence-similar antitags.
A pair of near complementary antitags (or tags) are—in sequence—very similar to the reverse complements of each other, so that a strong hybridization between them will occur, making a stable DNA duplex (e.g., the duplex 5′-ACTG-3′/5′-CAGG-3′). Alternatively, a pair of sequence-similar antitags are almost identical except for a few nucleotides (e.g., 5′-ACTG-3′ and 5′-ACTT-3′) and so are likely to bind each other's targets. For instance, 5′-ACTT-3′ is near complementary to the target of 5′-ACTG-3′ and is thus more likely to form a duplex with the wrong target as 5′-ACTT-3′/5′-CAGT-3′ (Fig. 1). Therefore, to achieve orthogonality, antitags are designed to be oligonucleotides that are most dissimilar from each other as well as from each other's complements. Below we further refine this criterion via 4 joint subsidiary problem classes.
I. It is undesirable to have highly stable DNA duplexes formed by antitag–antitag interactions, as might be the case with near-complementary tags. In other words, even when the targets are not present in the solution, we desire to have DNA probes to be able to stay in single-stranded (melted) forms at a particular temperature to insure the reliability of their identification, else the most adjacently placed identifiers would tend to bind to each other instead of their targets even when the samples are present—thus decreasing assay sensitivity. This problem is dealt with by designing the antitags to have the melting temperature of any oligonucleotide duplex formed by the distinct antitags as low as possible.
In particular, securing a low level of probe–probe interactions can offer greater flexibility in certain microarray design problems dealing with probe–probe affinities, such as border minimization problem (Hannenhalli et al., 2002) (i.e., minimizing overall border length of nucleotide masks in synchronous chip design), or in general microarray layout problem (de Carvalho and Rahmann, 2006) (i.e., arranging the probes on microarray chips by synthesizing the oligonucleotides on specific chip locations in the order which should maximize the overall quality of probe syntheses) (de Carvalho and Rahmann, 2007).
II. Tags are analogously optimized for reduced dimerization in assay solution to insure that the targets remain single-stranded within some temperature range and avoid dimer formation when incubated on the microarray.
III. The design problem is incorporated into the case of similar tags by keeping the melting temperature of any noncognate antitag–tag duplex as low as possible in order to prevent binding of probes to the wrong targets, which is known to be one of the main causes of cross-signal contamination (Ben-Dor et al., 2000).
IV. Finally, probes (antitags) that bind weakly to their targets (tags) must be eliminated. That is, probes must exhibit strong perfect-hybridizations with their targets, allowing for the existence of a separation temperature (“safety distance”) regime, where most perfect-hybrids are in duplex form while most cross-hybrids are still melted, thus allowing strong signal-to-noise ratio.
When optimizing binding specificity, we also take into account secondary structures of antitag/tag oligonucleotides, such as hairpins, bulges, interior loops, etc. (Zuker, 2003). This ensures that the melting temperature of a duplex is computed by simulating the self-folding of the strands as well as other strand structure formation and profiling their competition [i.e., between double-strand (hetero-dimer), self-folding (self-dimerization), and homo-dimer formations].
Using these criteria, we can now formulate the design problem for the construction of a tag/antitag library based on Tm optimization.
Melting temperature optimization
In the experimental settings, double-stranded DNA melting is most readily affected through temperature control, whereby the solution is heated until the stacking interactions and hydrogen bonds primarily responsible for holding the molecular duplex together are broken, resulting in its dissociation into two single strands (Strachan and Read, 2010). This feature could be efficiently utilized when performing highly specific DNA target recognition, if—for a given tag/antitag library—there exists a temperature range where all the desired perfect-hybrids are present in the predominantly duplex form, while all of the spurious cross-hybrids have largely melted.
The formation/dissociation of a duplex is a process of chemical reaction, where the transition from one state to another state results in a change of energy in the system. The change in Gibbs free energy (ΔG) of the reaction is equal to the thermodynamic transition parameters enthalpy (ΔH) and entropy (ΔS) as ΔG=ΔH−T×ΔS. Note that ΔG and hence the equilibrium constant of the hybridization reaction depend on the temperature of the surrounding environment. In this setting, given an equal mixture of complementary DNA strands, a “melting temperature” of the underlying sequence can be defined as the temperature at which half of the molecules are in a duplex and half in single-stranded form. Using a simple two state (hybridized vs dissociated) DNA strand model, melting temperature can be estimated as
where Ct corresponds to molar strand concentration, R is the universal gas constant, and f=1 when the two strands are the same or f=4 if the strands are different (Markham and Zuker, 2008). The sequence-dependent factors are applied as the amount of changes in transition parameters ΔG, ΔH, and ΔS, and salt corrections are added to Tm from published tables (Allawi and SantaLucia, 1997; SantaLucia, 1998; SantaLucia and Hicks, 2004).
However, the above equation is a very simple calculation of Tm that models double-stranded DNA dissociation as a single-step process. A more comprehensive definition of the “melting temperature” may be as the temperature at which the system passes through a sharp heat capacity maximum, ∂Cp/∂T=0, due to the cooperative loss of stacking interactions in the course of duplex dissociation (Mikulecky and Feig, 2006; Markham and Zuker, 2008). While this typically yield similar values to the ones obtained with the former half-melted duplex definition above (which also allows for a more direct interpretation of empirical results), the latter could be most immediately related to and calculated from the underlying biophysical and biochemical properties of the DNA polymer molecules involved.
In the latter case, the heat capacity is given by the equation (Mikulecky and Feig, 2006)
and Tm is obtained from the maximum or local maxima of the heat capacity curve such that
In this work, we used the Tm(Cp) calculation given in (2) as the melting temperature metric (Mikulecky and Feig, 2006; Markham and Zuker, 2008).
Problem formulation
We seek to construct a set of orthogonal DNA probes such that the difference (X) between the minimum Tm of perfect-hybridizations (antitag–tag) and the maximum Tm of cross-hybridizations (antitag–antitag, tag–tag, antitag–[foreign]tag, [foreign]antitag–tag) is as large as possible across any pairing of sequences in the collection. Namely,
1. For all combinations of antitags/tags of U and V:
where 2. Minimize X.
Notice that when X is a positive number the melting temperature of perfect- and cross-hybrids cannot be fully separated (Fig. 2); otherwise, when X is a negative number it quantifies the “safety distance” (separation temperature) between perfect- and cross-hybrids. In the latter case, X represents the size of the range from which a temperature could be selected to enable high fidelity identification assay to be performed (Fig. 3). Note that the more negative X is, the larger the safety distance from which a temperature could be chosen and so the more efficient the assay separating cross- from perfect-hybrids. Thus, in this sense, “−X” could be viewed as a global measure of orthogonality for any given set of oligonucleotides.

Heat map and histogram of the average random set, with orthogonality X=10. Color images available online at www.liebertpub.com/nat
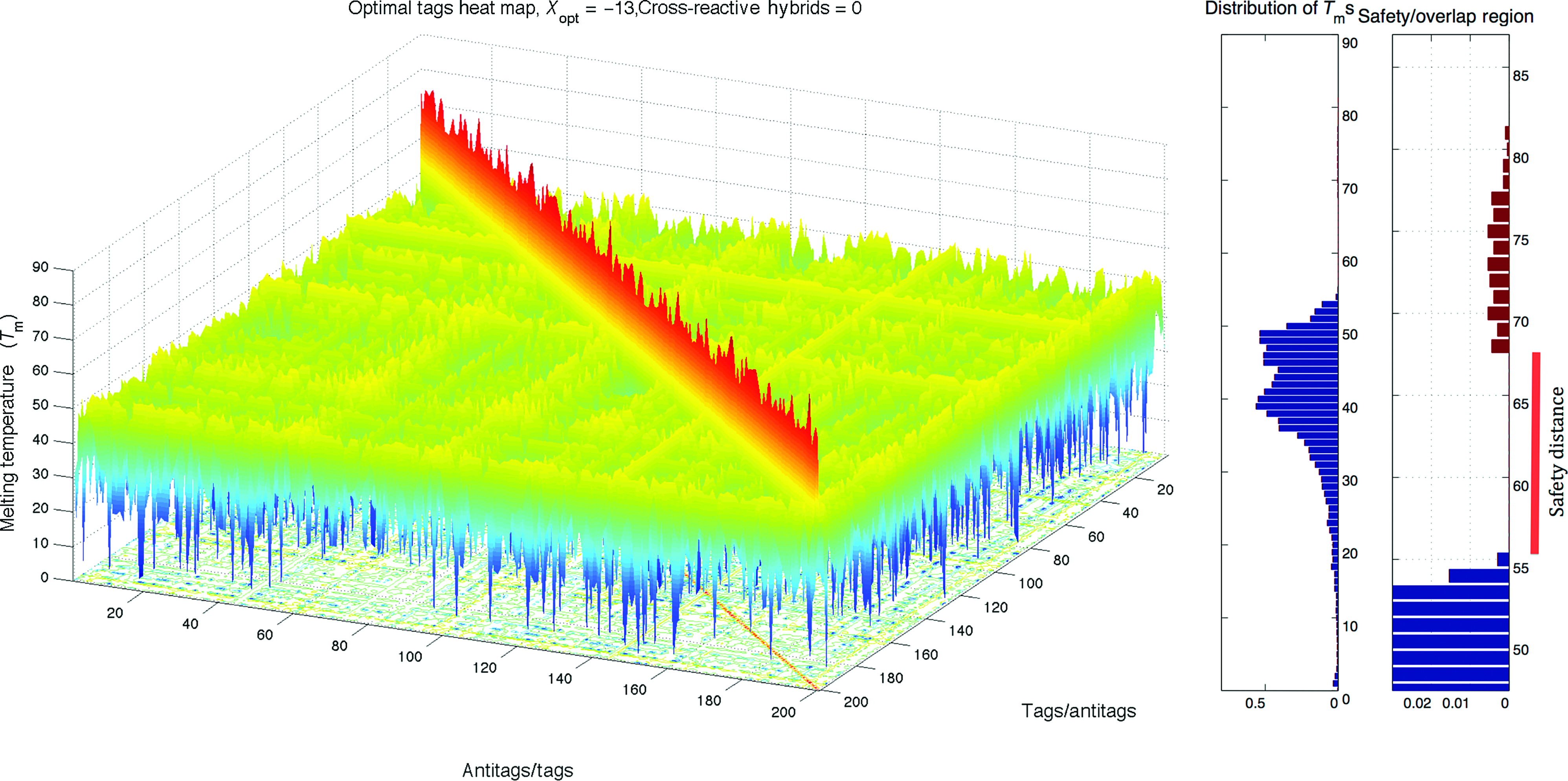
Heat map and histogram of the optimized Optimal Tags, X=−13. Color images available online at www.liebertpub.com/nat
Figures 3 and 4 show the histogram of melting temperatures for all possible perfect- and cross-hybrids in two TaT libraries. Both libraries have nonoverlapping temperature ranges between perfect- and cross-hybrids that allow for their full separation thus achieving orthogonality. These tag collections will later be discussed in further detail.
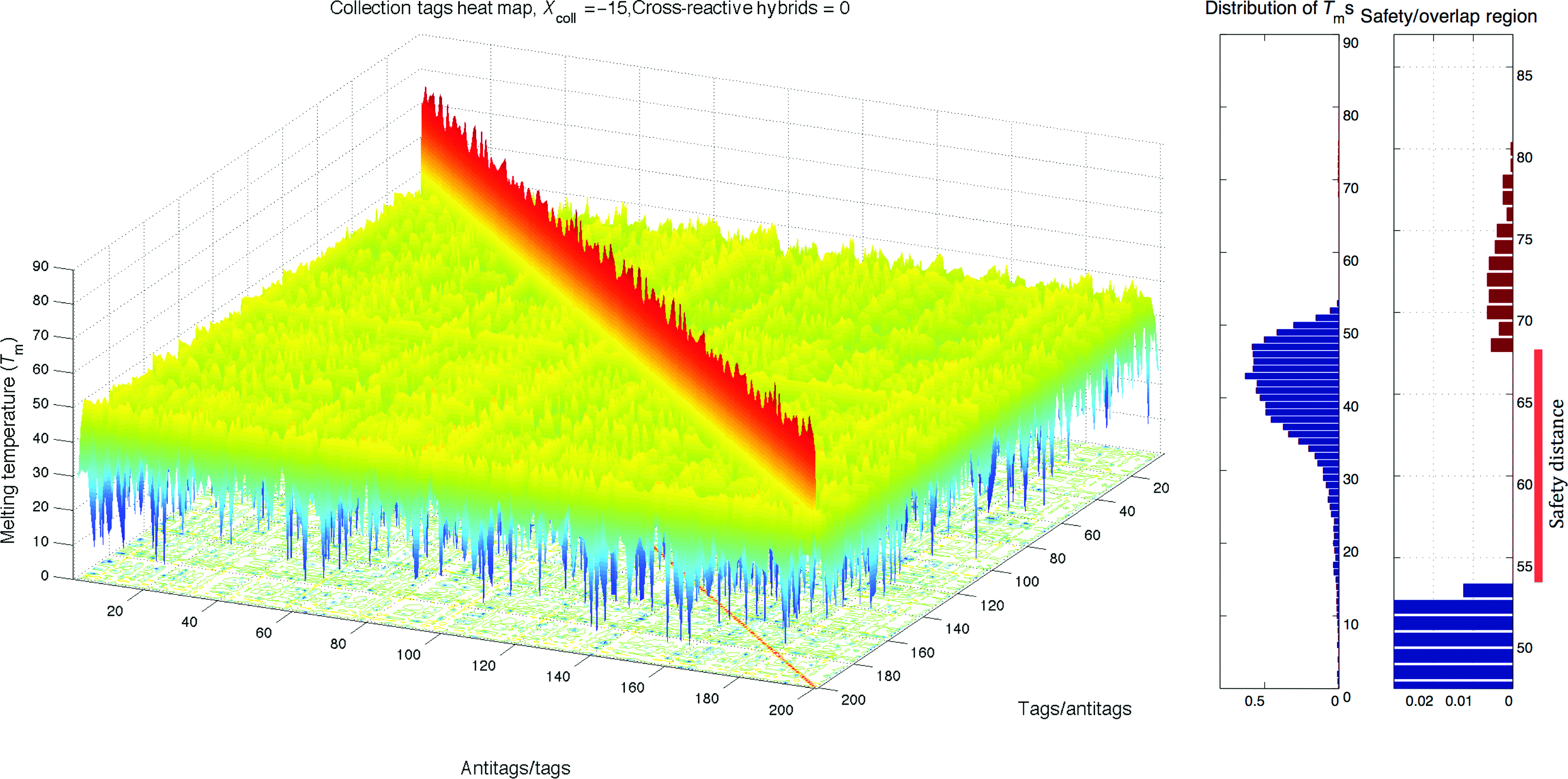
Heat map and histogram of the optimized Collection Tags, X=−15. Color images available online at www.liebertpub.com/nat
In this context, given a set of N unique L-mer probes, the diagonal elements of a 2N×2N matrix of hybridization scores (heat map) represent the perfect-hybridizations, while the off-diagonal elements represent the cross-hybridizations (Fig. 5). The orthogonality score X of the set is the outcome of the cost function associated with the heat map (i.e., the difference between the minimum diagonal and the maximum off-diagonal values). The corresponding probe set can be defined as a point in the sample space that consists of 4 N×L possible sequence sets.
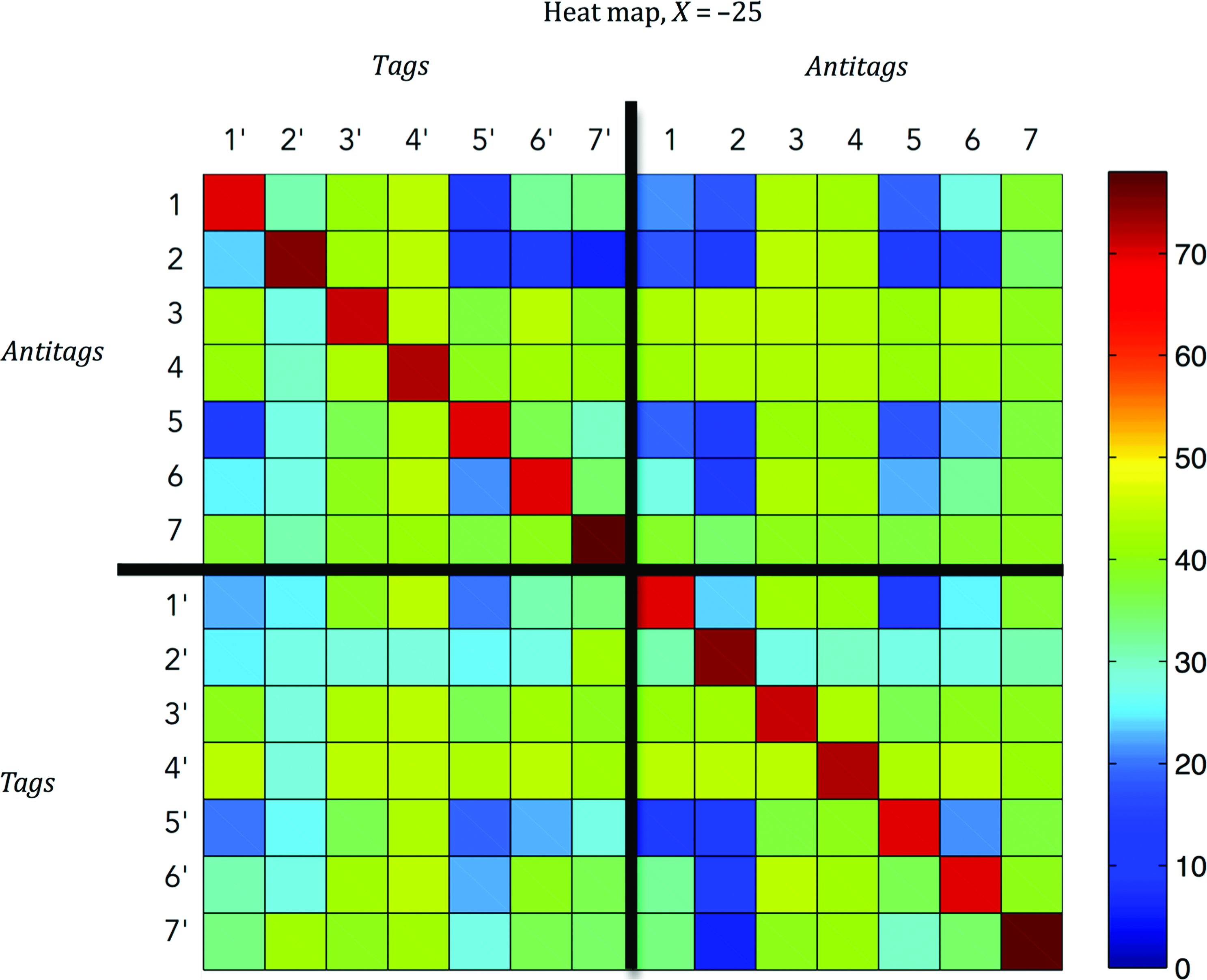
Heat map for a tag/antitag (TaT) library corresponding to 7 orthogonal probes. Upper-left 7×7 matrix represents the melting temperatures (Tms) of possible tag–antitag bindings; here, probe–target bindings (diagonal) show strong (red) perfect-hybridizations while probe–[foreign]target bindings (off-diagonal) have weak (blue) cross-hybridizations. Upper-right 7×7 matrix elements represent the probe–probe cross-hybrids. Lower-left matrix shows the target–target bindings. Lower-right matrix shows the target–probe (diagonal) and [foreign]target-probe (off-diagonal) bindings, which is the transpose of the upper-left matrix. Color images available online at www.liebertpub.com/nat
To improve the orthogonality X of a set of antitags one could perform an optimization by walking over the sample space of sets one by one, and searching the entire space by “brute force.” However, for most practical applications, antitag length L and set size N are large enough to make such a walk over all possible sets computationally infeasible. Furthermore, replacing an antitag once causes a transition to another point/set in the sample space, which affects not only the replaced antitag's perfect-hybrids but also those of all cross-hybrids related to it (i.e., 4N−1 cross-hybrids). Replacement of any one antitag generally affects the cross-hybrid affinities of all other antitags in the set. To see this, let us assume that a subset of antitags set* in the current set (set* ⊆ set) has been optimized (i.e., set* corresponds to the weakest cross-hybrids and the strongest perfect-hybrids). Should the next step of the optimization, however, attempt to replace an antitag in set\set* with a new antitag, which happens to diminish the orthogonality of set*, for example, because new antitag is near complementary (or sequence-similar) to one in set*, the overall set orthogonality is likely to be diminished and set* would need to be reoptimized as long as new antitag remains in the set. Because of this joint nonlocal nature of the problem, the optimization has to account for all interactions in the set at once in each step. Thus, no single oligonucleotide, subset of oligonucleotides, or even nucleotide subsequence can be a priori guaranteed to remain optimal or suboptimal with respect to a specific orthogonal TaT library design. That is, if we divide the problem into subsidiary problems and optimize them disjointly (i.e., considering only a group of antitags at a time), the complete solution may only result in a limited orthogonality improvement.
As an example, consider the sub-matrixes of the simple heat-map given in Fig. 5 and the four subsidiary problem classes defined previously, and let us attempt a method that solves these subsidiary problems separately using some sort of a “greedy” approach (e.g., Xu et al., 2009a). That is, suppose that we have first determined a set that has the weakest antitag–antitag interactions as measured by low Tm levels, a second set that has the weakest tag–tag interactions, a third set that has weakest antitag–[foreign]tag interactions, and a fourth set that has strongest antitag–tag interactions with high Tm scores. Then, it may be expected that the larger the set size, the fewer antitags these 4 sets will have in common. So, despite the easy implementation and faster convergence in each problem class, the ultimate solution will not be the global optimum.
Because of this, using such divide-and-conquer heuristics may not be the best strategy when looking for the global optima. Instead, we need to formulate a method that can be iteratively applied to generate simultaneous improvements across all interaction classes in tag/antitag library.
Designing orthogonal tags by probe elimination
To make this more concrete, consider a recent work by Xu et al. (Xu et al., 2009a), which describes a framework for elimination of unsuitable probes from a large initial set. It consists of 2 elimination phases: the first is based on analyzing nucleotide contents of sequences individually; and the second evaluates the mutual similarity between pairs. The method starts with a randomly generated 10 million 25-mer oligo DNA sequences. The next step involves the elimination of sequences by using a number of filters: restriction enzyme filter for excluding the candidates that contain restriction enzyme sites reserved for cloning purposes, Tm filter based on the nearest neighbor (NN) model for excluding sequences that have perfect-hybrid Tms that are too high (>68°C) or too low (<58°C), GC composition filter for excluding sequences with GC contents above 60% or below 40%, and repetitive sequence filter for excluding sequences that contain 5 or more single nucleotide repeats or 4 or more double nucleotide repeats, respectively. Sequence candidates that pass all of these filters are then compared among each other for sequence similarity using basic local alignment search tool (BLAST). Finally, two distinct sets of 240,000 orthogonal sequences are determined by the Network Elimination Algorithm (Xu et al., 2009b).
This protocol can provide a fast approach for designing large sets of orthogonal sequences, but the speed is attained at the cost of reduced orthogonality, since filters employing probe design rules have to unnecessarily restrain the variety and bias the selection of sequences because many fundamentals of DNA hybridization cannot be “boiled down” or well understood in terms of such simple selection rules. For example, in (Xu et al., 2009a) it is shown that the Tm filter based on the NN model is not very helpful as a probe design rule due to frequent occurrences of good probes with Tms falling out of the minimum-maximum Tm interval that end up being filtered out. Additionally, reliance on BLAST ignores the physical differences (e.g., energetics of binding) between different sequences, since considering pair-wise alignment is not sufficient to establish physical properties, such as melting temperature, which are frequently the ones most relevant in practice. Besides, there are at least 100 million other sets of 10 million distinct sequences beyond those allowed by the filtering constraints, since the sample space for 25-mers is as large as 425≅11×1014. Although the restricted set of 10 million sequences randomly generated in the initial step may appear large enough for determining 240,000 orthogonal tags, the contribution from the universal sample space is likely to contain many more orthogonal solutions.
Another related work presented by Shoemaker et al. (Shoemaker et al., 1996) had proposed a quantitative method for generating distant 20-bp tags, which have since been widely used in a number of studies. This method is based on 3 elimination phases. First it begins by considering all 20-mer sequences in the sample space, then it performs the elimination based on known hybridization problems, such as secondary structure, base composition, runs of single nucleotides, etc. Next, it uses pair-wise analyses to eliminate sequences that contain common 9-mers by assuming that long contiguous base-pairings (≥10 bp) is what largely causes cross-hybridization. The resulting 51,082 distinct sequences are predicted to have similar Tms for perfect-hybrids within a min–max interval. Finally, the remaining sequences are passed through another filter to further eliminate those that are most likely to cross-hybridize, resulting in sets of different sizes—9,105, 2,643, 853, 170, and 42 tags—depending on threshold levels.
The utilization of sequence-similarity and Tm filters proposed in both studies can allow fast searches for target sets of sequences; however, they are often inclined to unnecessary removal of good candidates as pointed out in Xu et al., 2009a. Such Tm filters evaluate thermodynamic characteristics of probes individually, while ignoring potential interactions with other sequences. A recent case study has further highlighted similar concerns for other aforementioned design filters by evaluating the probe collections from 74 Affymetrix arrays (Tulpan, 2012). In contrast, the orthogonality measure X proposed here is influenced by all types of interactions across tag/antitag library, thus potentially giving a more practical handle for limiting cross-hybridization. Furthermore, X is a measure less sensitive to probe nucleotide contents than that imposed by Tm filters.
Given these considerations, we propose a method that uses metric X for probe interactions in order to reduce the melting temperatures of cross-hybrids as much as possible, while securing high melting temperatures of perfect-hybrids, thus allowing for a broad separation regime (where a reaction run at temperature above maximum cross-hybrid Tm and below minimum perfect-hybrid Tm will produce predominantly true probe–target duplexes, while most cross-hybrids remain melted). Our problem formulation naturally corresponds to sequence-similarity filter and Tm filter, since minimizing X also minimizes problematic sequence similarities by reducing cross-reactivity between probes and forces the perfect-hybrid Tms to fall into narrow higher temperatures ranges. However, we still apply other probe design rules since they are widely accepted in literature for securing probe–target specificity (i.e., GC composition filter) and single and double nucleotide repetitive filters.
Designing orthogonal tags by simulated annealing optimization
When designing orthogonal tags (antitags), each with length L, the optimum set of N tags is the minimum point of the cost function over the N×L dimensional space S that contains 4N×L constellation points—each representing a probe set. The cost function of every point gives the outcome value of orthogonality metric, X. The sets of unique tags form a subspace U⊂S. Each of the
Using an iterative improvement strategy by doing a random walk in U leads the walk through a local minimum depending on the starting point that, however, may or may not be globally optimal. Since an iterative improvement always takes a path downhill (towards a more orthogonal set), it avoids transfers to higher points (less orthogonal sets). Yet, such points may potentially stand on the path toward the global minima and so a search method should be allowed to sometimes transfer to higher points. An approach based on simulated annealing optimization offers some appropriate schedules for allowing such hill-climbing transitions, thus avoiding being trapped in local optima. As employed in this study, the major benefit of the simulated annealing approach lies in the fact that the global optimum set may possibly be hidden behind some sequences that are excluded in the initial sequence pool or filtered by the individual constraints such as Tm filter based on the NN model used in other algorithms. Although these filters usually find problematic sequences, excluding them should be avoided as much as possible. That is, some points/sets may not be very orthogonal but can still act as a narrow gate on the way towards the global optimum set; thereby, visits to these points are essential when searching for global optimum and should not be neglected due to pool or sequence constraints in an optimization procedure. When compared with other optimization techniques, another benefit of simulated annealing is its greater scalability with increasing number of sequences.
The idea of simulated annealing for the combinatorial optimization was introduced by Kirkpatrick et al. (Kirkpatrick et al., 1983). In physics, the annealing process of a solid material starts with an initial temperature level, C, which is allowed to gradually decrease, eventually reaching thermal equilibrium. Based upon this analogy, the simulated annealing optimization approach used here starts with an arbitrary state that is a random set of unique tags. The state of the process is defined by the measure of orthogonality, X, of the current set. During each iteration, a tag is replaced by a randomly determined unique tag, which is called a “new state” and the set with the new tag is called a “neighboring set.” By replacing one tag at a time, the algorithm checks the orthogonality of every new state and decides whether or not to transfer to the neighboring set with a gradually decreasing preference.
In each step, the probability of transferring to the new neighboring set depends on the closeness of the new state, iteration/step number, k, and the annealing temperature constant, C, as:
where
Note that if a better or an identical score is achieved, the transfer is realized with probability 1 (notice that [Xnew−X]+ function in PT takes the maximum of 0 and the gain in score); if the score is worse – there is still a possibility of transferring depending on the closeness of the new state's score and the annealing temperature Tk—an inversely proportional logarithmic function of the iteration number, k. This decision of transferring allows for the “hill-climbing” moves that prevent the process from being stuck in a local minima. Notice that the probability of transferring PT is logarithmically decreasing as the optimization continues, since Tk is a logarithmic function of k. Early steps of the process are also more unstable because of high Tk values so transitions from one state to another may be frequently observed in similarity to the physical annealing. Thus, the algorithm more likely allows for the hill-climbing moves in the early phases of optimization. These hill-climbing moves allow the process to walk over a very scattered subspace of U, increasing the possibility of finding a path to the global optima. Later, the process reaches an equilibrium level so that there remains no better neighboring set to which to transfer. This is the result of PT being lowered to very low percentages, so that it rejects almost any hill climbing move and the optimization goes through a stable phase. In this later phase of optimization, the process reaches a local minimum, which is, with high probability, the global minimum in the coordinate space U that will also be the global minimum in S.
Most authors studying simulated annealing have concentrated on determining an ideal annealing schedule, basically defining an initial temperature C and a convergence condition to finish the optimization process (Hajek, 1988). In this work, we assumed that the annealing iterations are large enough until PT drops to the threshold of 0.01. For a small set (tags<20) of 20-mers, most of the optimization runs are assumed to end up with global optimum solutions when C=2, since the same process with C=3 does not achieve any better result. For large libraries (tags>100) and/or libraries with longer sequences (>50-mers), however, it is expected that the process is more likely to reach the global optimum when C≥2. The details of simulated annealing optimization method are summarized below.
Summary of the algorithm
• Initialize the algorithm.
- Pick a random initial point in U: Define an initial random dataset set consisting only of unique tags. - Generate the heat map for the set and compute the orthogonality score as X=max[Tm,cross(set)] – min[Tm,perfect(set)]. - Initialize the best point of the random walk in U. setbest←set Xbest←X
• Iterate until convergence: PT<threshold.
- Do a random walk to a new neighboring set setnew by replacing a random tag and compute the new score Xnew after updating Tms of perfect and cross-hybrids related to the replaced tags on the heat map. - Decide on the transition: * Transfer if PT>U (0, 1), where U (0, 1) is a random number uniformly distributed on [0, 1]. set←setnew X←Xnew
* Update the best set if X<Xbest. setbest←set Xbest←X - k←k+1
• Return setbest and Xbest.
• Replace the tags that are not approved by probe design rules.
• Mark as • Re-optimize setbest by (mostly) searching the neighboring sets of * Initialize the starting point with {setbest, Xbest}. * Iterate the optimization procedure until convergence:
• Return setbest and Xbest.
Materials and Methods
Complexity
The proposed algorithm computes the heat map of an initial set in O(N2) time by simulating all N×(2N+1) perfect and cross-hybrids. Later in each step, the heat map is updated for the replaced tag in O(N) time that requires the simulation of 4N−1 putative hybrids. If the heat map is known, the optimization problem reduces to polynomial time complexity, O(Nk), where k is the number of iterations. The proposed method is applicable to parallel processing (i.e., in each step, 4N−1 simulations can be distributed among available processors of a cluster for the speedup).
The Tm of each duplex is computed after generating the duplex's melting profile by UNAFold software. This dependency may represent a major computational speed bottleneck in practical implementation, since each step requires pipelining the results from UNAFold to continue the optimization process. However, UNAFold is a greater source of accuracy in various hybridization conditions since it is based on such underlying physical properties of tags as nucleic acid folding structures, homo-/hetero-dimer formations and melting profiles, etc. (Markham and Zuker, 2008).
UNAFold
The hybridization of each pair of tags and/or antitags is simulated by UNAFold and the corresponding melting profile is generated. Given the melting profile of a duplex, there are various ways of computing its melting temperature. In our optimizer software, we considered the Tm(Cp), the prediction of melting temperature based on the heat capacity. In case of multiple distinct peaks on the heat-capacity curve, the Tm(Cp) value corresponding to the largest temperature is assumed as the melting temperature (Markham and Zuker, 2005). We used the following parameters as standard in all Tm computations:
• Initial concentrations: uniform concentrations are selected as 10 μM for each tag.
• Salt conditions: Na+=25 mM, Mg++=10 mM for every hybridization.
Orthogonal tag optimizer server
A user-specified set of DNA tags can be requested from the online implementation of the proposed method (www.columbia.edu/∼ae2321/TagApplet/starter.html).
Users can submit a job by defining several inputs for the dataset of interest:
• Initial concentrations (mol/L): The uniform strand concentration for each tag.
• Salt conditions (mol/L): Na+ and Mg++ concentrations in the solution.
• Number of tags, N: A set of 1,000 tags can be optimized approximately in 3 weeks of processing on a single processor.
• Tag length, L: Flexible for different sizes.
• Number of simulations: Several optimizations can be run with different initial points and the best result will be presented.
• Optimization parameter, C: Annealing temperature constant to adjust optimization schedules.
• E-mail address: When the results are ready, user is notified via email.
Results
In (Oh et al., 2010), a universal TagModule Collection is introduced for parallel genetic analysis of microorganisms. This collection has 4,280 TagModules. All TagModules are identical except for two unique 20-mer DNA identifiers labeled as Uptag and Downtag. These identifier tags have been selected from the sets generated in Shoemaker's works (Shoemaker et al., 1996). Here we are interested in the orthogonality of these 8,560 Uptags and Downtags in comparison with the 20-mer tags optimized by our approach.
Recall that the expected orthogonality for a set of probes reduces as the number of probes in the set increases. Although the library of 8,560 DNA probes has certain orthogonality with respect to its size, it is not guaranteed for smaller sub-sets to have the same relative orthogonality. We evaluated the degree of interactions X among tags in TagModule collection by randomly determining a set of 100 tags and generating its heat map. By repeating this, 30 different random sets were drawn, and it is observed that in almost every set there is a cross-hybrid contamination, where the corresponding orthogonality scores range from X=−1 to X=55, with the average score of Xavg=9.9. Figures 2, 6, and 7 show the heat maps and distributions for several sets among 30 random sets: an average set with X=10, the best random set with slight orthogonality X=−1, and the most contaminated set with X=55, respectively. As can be seen, the subsets of TagModule collection contain highly cross-reactive probes and thereby, an optimization procedure should be applied to discover orthogonal solutions. We applied our method to perform this optimization task since it studies all mutual interactions among tags.
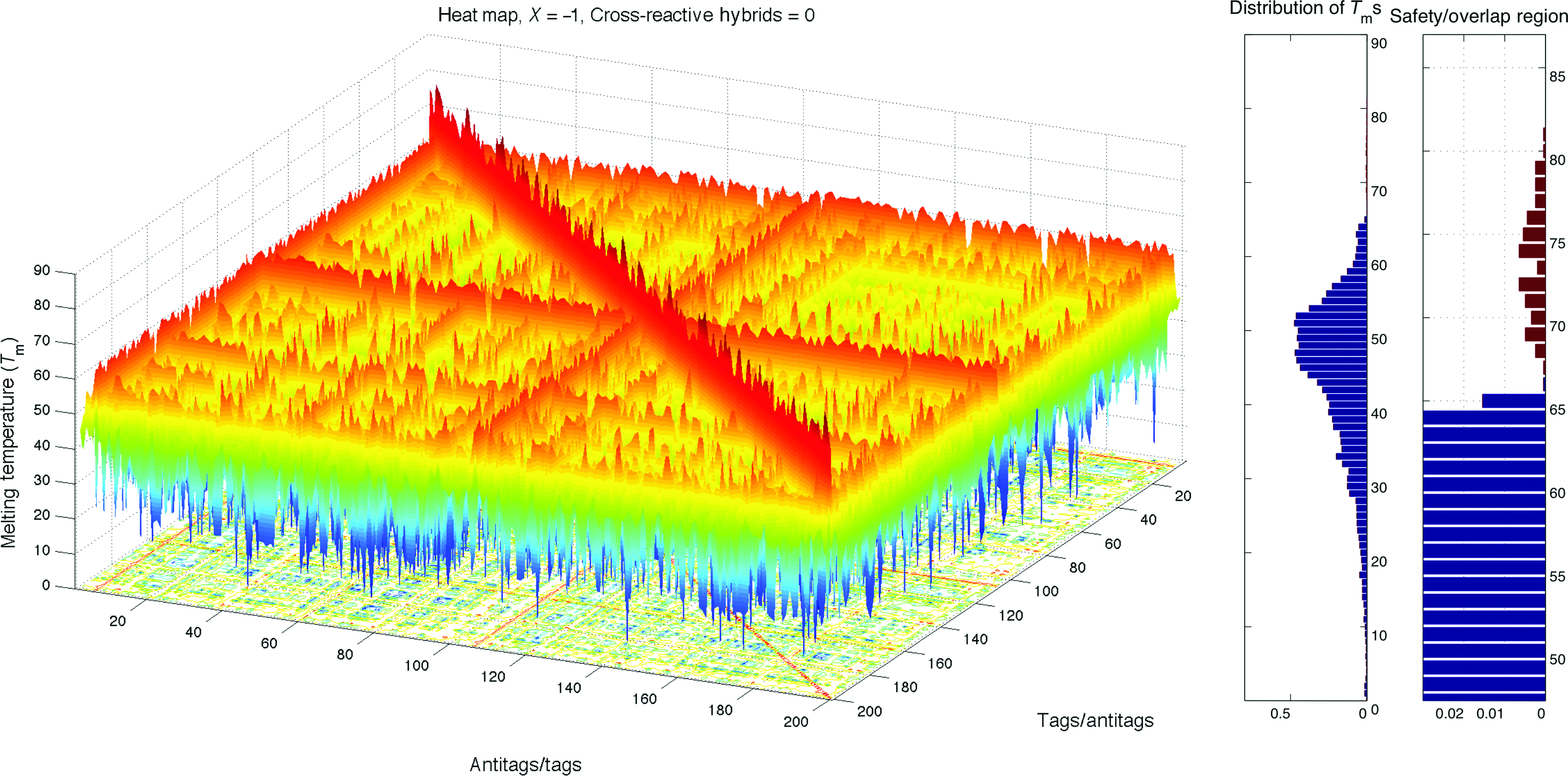
Heat map and histogram of the best random set, X=−1. Color images available online at www.liebertpub.com/nat
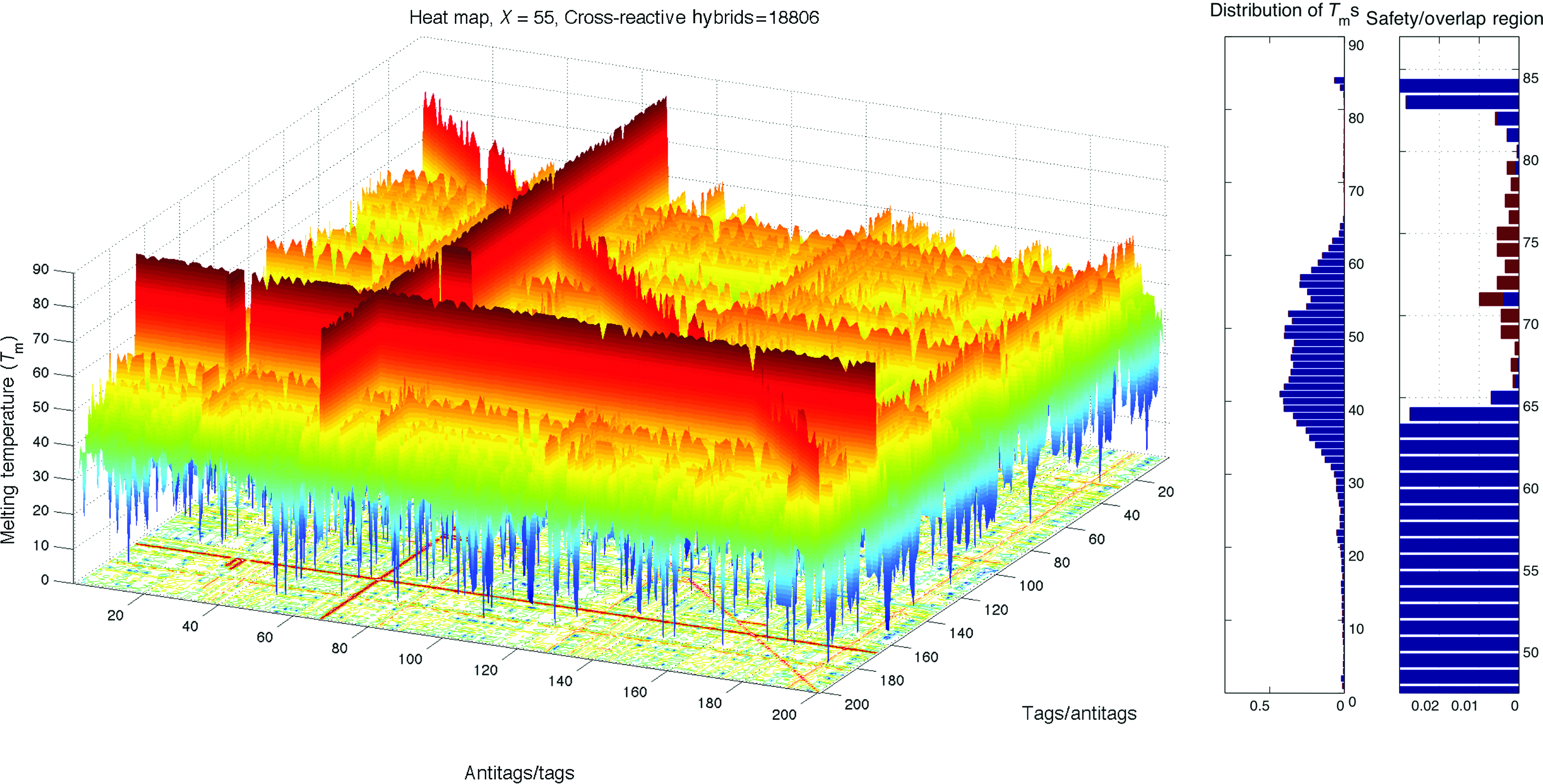
Heat map and histogram of the most contaminated random set, X=55. Color images available online at www.liebertpub.com/nat
By using simulated annealing optimization, we have elucidated the most mutually orthogonal subset of 100 sequences from the TagModule collection. That is, we have restricted the space of possible sets to those only consisting of given 8,650 probes and used an appropriate annealing schedule (C=2) for the optimization. We simulated 30 parallel runs and extracted the best data set, Collection Tags, corresponding to the lowest score of degree of interaction, XColl=−15, which is, with high probability, the global solution in TagModule collection for the given set size. Figure 4 shows that cross-hybrid Tms are distributed from 0°C to 53°C, and perfect-hybrid Tms are distributed from 68°C to 80°C. The improvement is obvious when compared to the orthogonality of random subsets (Figs. 2, 6, 7) of the TagModule collection.
For comparison, we have applied our approach to the same problem—using the same optimization schedule—with the only difference being that no quantitative filters are used other than eliminating identical or complementary tags and finally replacing those that are not approved by the given probe design rules. Each optimization is executed by drawing purely random DNA tags and performing the pair-wise analyses for each type of tag/antitag interaction by evaluating full thermodynamic properties of pairs, and replacing undesired probes that cause strong cross-hybridizations and/or weak perfect-hybridizations. Among the 30 optimized sets, the best one, Optimal Tags, with the lowest score of degree of interaction XOpt=−13 is displayed in Fig. 3. In this set, cross-hybrid Tms are distributed from 0°C to 55°C, and perfect-hybrid Tms are distributed from 68°C to 81°C.
Note that our Optimal Tags set reaches the same degree of cross-interactions on median (41°C), and in both sets the perfect-hybrids are distributed narrowly in a similar range and reduce to the same lower bound (Tm=68°C). Although Collection Tags has slightly better orthogonality score than Optimal Tags, this can be explained by the faster convergence of algorithm resulting from the restriction of optimization to the TagModule probes. Consider further that it is very unlikely to elucidate the Optimal Tags from a given database such as TagModule collection or even from large random pools. That is, after several tests we observed that none of the probes in Optimal Tag Collection are found in randomly generated initial pools consisting of 1 million 20-mers. This may indicate the diminishing effect of restriction of the search to such initial pools. Notice that by using a pool of 1 million≃410 distinct 20-mers one can only cover the sample space of a 10-mer sequence, which is equivalent to predefining half of the nucleotides randomly in each elucidated probe.
Discussion
In this work, we propose an approach for designing orthogonal sets of probe–target oligonucleotides. The method offers potentially enhanced specificity and reduced cross-reactivity of tag–antitag recognition because it does not rely on auxiliary constraints—such as sequence-similarity filters, Tm filters, restricted oligonucleotide databases or predefined initial pools—which may restrict other approaches to potentially suboptimal solutions. The proposed method offers an alternative way of determining orthogonal probes by searching an entire feasible set on the sequence space so that the elucidated set can optimally satisfy any predefined properties as far as possible. On the other hand, if it is necessary to rely on a given probe database/source sequences (e.g., when designing polymerase chain reaction primers), our approach can be used to find, with high probability, the global optimum solution for the given database (e.g., the most mutually orthogonal subset of a given size —Collection Tags—from the TagModule collection designed previously (Oh et al., 2010).
The key benefit of our approach is the additional degree of freedom it offers towards the selection of tag/antitag sequences, which reach the global optimum, while imposing a thermodynamic quality control feature that evaluates all probe and target interactions involved. This degree of freedom helps the algorithm search a very scattered sample space as well as deal with similar and near-complementary tags that have led to major drawbacks in TaT design techniques (Tulpan, 2012). The proposed approach employs the simulated annealing method in order to free the optimization process from being stuck in local minima, finding—with high probability—the global solution of a probe set with the desired properties. These properties can include various individual or pair-wise constraints and/or probe design rules imposed on the tags, such as, restricting the tags in order to exclude/contain restricted enzyme sites or other sequence patterns or motifs, eliminating tags that have consecutive base-pair matches, defining the range of allowed GC contents, etc.
Here, we used the difference between the minimal melting temperature of perfect oligonucleotide hybrids and the maximal melting temperature of any spurious self- or cross-hybrids as the optimization parameter. What makes melting temperature between interacting probes and targets a practically relevant quantity when measuring orthogonality is that Tm values can be both controlled in situ as well as robustly estimated by accounting for the physical properties of the tags. Note that this behavior of DNA hybridization cannot be predicted by only studying the nucleotide matches between sequence pairs. In this work, we have employed the software published by (Markham and Zuker, 2008) for generating the melting profiles of each putative hybrid. For a sophisticated analysis of hybridization behaviors, it combines a number of auxiliary programs that performs the free energy minimization and full partition function calculations and stochastic sampling in hybridized or folded sequences (Hofacker, 2003; Zuker, 2003; Ding et al., 2004). For the interacting molecules of a duplex, the competitions between all possible molecular species are taken into account, (i.e., a hetero-dimer duplex, 2 homo-dimers, and 2 foldings). The folding predictions are based on free energy minimization and full partition function calculations, and all possible competitions between the dimer formations and foldings of the individual strands are simulated. For each species, the partition function computations are performed over a range of temperatures with overall ensemble quantities being computed for ΔG, ΔH, ΔS, and heat capacity after combining the results (Markham and Zuker, 2008).
It should be noted, however, that these calculations might still fall short in accurately estimating Tm because the algorithms do not always detect some unusually stable structures in single strands (Zuker, 1989). Furthermore, the employed software is designed for simulating the hybridizations in solution. If the probes are being designed for microarrays, it is necessary to use the effective concentrations of the oligos that will be immobilized on the microarray surface (Markham and Zuker, 2008). That is, one needs to compute the surface phase concentration and solution volume per surface area and to calculate the effective concentrations (Zhang et al., 2005). This calculation may be complicated due to certain factors that cause the effective volume to differ from the calculated ratio of total volume to total surface area (Zhang et al., 2005). In surface hybridizations, another important factor is the diffusion effects that determine the rate of hybridization (i.e., the time needed to reach the equilibrium state in a hybridization cell) (Markham and Zuker, 2008). The rates of hybridization in microarray cells can vary depending on the binding site concentrations and the equilibrium binding constants of the duplexes being hybridized—for instance, cells with the most stable duplexes may take longer to reach the equilibrium state than may otherwise be expected (Livshits and Mirzabekov, 1996). Even with incubation times on the order of hours, some sequences may not reach equilibrium in their cells and so probe–target signal strengths may vary due to the differences in hybridization rates. Thus, it should be emphasized that a probe set being designed by the proposed method may deviate from the expected characteristics due to aforementioned issues.
Hybridization characteristics of an oligo duplex also depend on various environment factors (e.g., salt conditions, oligo concentrations, etc.). It is well known that the melting temperature of an oligonucleotide or polymer hybrid is a salt dependent metric, whereby Tm decreases with lowering of salt concentration (Schildkraut and Lifson, 1965). To account for with this, the salt correction formulae published by (SantaLucia, 1998) are used to refine the thermodynamic parameters ΔG, ΔH, and ΔS when generating the melting profiles. The employed software further allows for ranges of salt conditions: the sodium concentration [Na+] must be between 0.01 M and 1 M, and magnesium concentrations [Mg2+] must be lower than 0.1 M in default oligomer mode (Markham and Zuker, 2005). This can give flexibility for the Tm predictions when optimizing oligonucleotide set designs under specific conditions.
The proposed method is based on simulating every hybrid of an initial probe set to generate the initial heat map and then simulating the putative hybrids of the tags being replaced during the optimization. The number of possible interactions in a probe library grows exponentially as the number of tags to be designed increases, so generation of the initial heat map also increases computational costs. Although this can be mitigated by applying parallel processing, the dependence on the software that generates the melting profiles of hybrids poses another limitation. Thus, it may be necessary to define a realistic maximum for the size of the library that can be designed using the proposed method in each individual setting. In our tests, applying parallel processing in combination with the appropriate annealing schedules, the proposed method can optimize sets that contain tens of thousands of sequences, for example, 10,000 tags can be elucidated in 8 days of processing on a cluster of 1,000 central processing units.
Footnotes
Acknowledgments
XW and MS conceived of the project. AE, GJ, XW, and MS participated in the design of the method. AE performed the computer experiments; AE and MS contributed in the writing of the draft. All authors read and approved the final manuscript.
Author Disclosure Statement
No competing financial interests exist.