
Abstract
Expansions of CUG trinucleotide sequences in RNA transcripts provide the basis for toxic RNA gain-of-function that leads to detrimental changes in RNA metabolism. A CTG repeat element normally resides in the 3′ untranslated region of the dystrophia myotonica-protein kinase (DMPK) gene, but when expanded it is the genetic lesion of myotonic dystrophy type 1 (DM1), a hereditary neuromuscular disease. The pathogenic DMPK transcript containing the CUG expansion is retained in ribonuclear foci as part of a complex with RNA-binding proteins such as muscleblind-like 1 (MBNL1), resulting in aberrant splicing of numerous RNA transcripts and consequent physiological abnormalities including myotonia. Herein, we demonstrate molecular and physiological amelioration of the toxic effects of mutant RNA in the HSALR mouse model of DM1 by systemic administration of peptide-linked morpholino (PPMO) antisense oligonucleotides bearing a CAG repeat sequence. Intravenous administration of PPMO conjugates to HSALR mice led to redistribution of Mbnl1 protein in myonuclei and corrections in abnormal RNA splicing. Additionally, myotonia was completely eliminated in PPMO-treated HSALR mice. These studies provide proof of concept that neutralization of RNA toxicity by systemic delivery of antisense oligonucleotides that target the CUG repeat is an effective therapeutic approach for treating the skeletal muscle aspects of DM1 pathology.
Introduction
RNA transcripts such as DMPK that contain a large number of CUG repeats acquire gain-of-function properties and promote RNA-mediated toxicity in cells and tissues expressing the mutant DMPK transcript (Wheeler and Thornton, 2007). The pathogenic transcript is retained in the nucleus (Taneja et al., 1995), where it entraps RNA-binding proteins such as muscleblind-like 1 (MBNL1) (Mankodi et al., 2001; Mankodi et al., 2003); poly (CUG) RNA also increases steady-state levels of hyperphosphorylated CUG-BP1 (Timchenko et al., 2001; Kuyumcu-Martinez et al., 2007). Both sequestration of MBNL1 and an increase in CUG-BP1 activity lead to abnormal splicing of a large number of RNA transcripts. Of note is the defective RNA splicing of chloride channel messenger mRNA (ClC-1 mRNA) (Mankodi et al., 2002), which has been shown to directly result in myotonia (Wheeler et al., 2007). In addition to abnormal RNA splicing, a consequence of mutant DMPK expression includes a remodeling of the skeletal muscle transcriptome (Osborne et al., 2009). Protein products with altered primary sequences resulting from abnormal RNA splicing and the dysregulated mRNA levels comprising the altered transcriptome would be expected to have associations and even causal relationships with specific aspects of DM1 disease.
There are currently no therapeutic agents in the clinic for DM1 that target toxic RNA, the primary pathogenic driver of the disease. Standard-of-care therapies for DM1 are mainly supportive and aimed at managing specific symptoms (e.g. myotonia) (Logigian et al., 2010). Pre-clinical evaluation of novel therapeutic approaches have been conducted in the HSALR transgenic mouse model that contains a human skeletal actin transgene harboring a 250 CTG trinucleotide insertion in the 3′ untranslated region. The HSALR model demonstrates several features of DM1 including Mbnl1 sequestration by CUG RNA and the ensuing RNA splicing abnormalities, alterations in the muscle transcriptome, and physiological aberrations such as myotonia (Osborne et al., 2009; Mankodi et al., 2000). Novel therapeutic modalities tested in this DM1 transgenic mouse model include small molecule ligands and antisense oligonucleotides (ASOs) that are designed to interact with CUG repeat RNA and liberate foci-associated Mbnl1 protein (Warf et al., 2009; Wheeler et al., 2009; Ofori et al., 2012; Parkesh et al., 2012). Degradation of the toxic HSALR transcript has also been evaluated by systemic administration of ASOs that hybridize to HSALR mRNA and invoke cleavage of ASO-targeted transcripts via ribonuclease H (RNaseH) (Wheeler et al., 2012). These ASOs contained a core of unmodified deoxyribonucleotides to enable RNaseH activity and were flanked on each end by 2′-O-methoxyethyl (2′-MOE)-modified nucleotides (Wheeler et al., 2012). Systemic administration of these 2′-MOE, RNaseH-active ASOs resulted in marked degradation of HSALR transcripts in skeletal muscle and produced a robust and sustained correction of the DM1-like phenotype in HSALR transgenic mice (Wheeler et al., 2012).
Three additional ASO approaches that specifically target the CUG repeat tract have been assessed in transgenic mouse models of DM1 (Mulders et al., 2009; Wheeler et al., 2009; Lee et al., 2012). Wheeler and colleagues demonstrated local correction of DM1 pathology in tibialis anterior (TA) muscle of HSALR mice subjected to intramuscular (IM) injection of a 25-mer morpholino oligonucleotide of CAG sequence (CAG25) (Wheeler et al., 2009). CAG25-treated TA muscles showed a decrease in the presence of ribonuclear foci, redistribution of Mbnl1 protein, correction of abnormal RNA splicing, restoration of ClC-1 protein expression and function, and reduction of myotonia (Wheeler et al., 2009). Local injection of CAG25 into TA muscle also led to a reduction of steady-state levels of HSALR transgene mRNA (Wheeler et al., 2009). Because morpholino oligomers are unable to activate the RNaseH pathway, this effect was attributed to enhanced decay of HSALR CUG transcripts, perhaps secondary to accumulation of these transcripts in the cytoplasm (Wheeler et al., 2009). Since the level of remaining HSALR transgene mRNA in CAG25-treated mice did not fall below a level sufficient to evoke the DM1 phenotype, the mode of action of CAG25 in vivo likely involved displacement of Mbnl1 protein from CUG repeat RNA (Wheeler et al., 2009). Although this localized therapeutic approach showed corrections in the DM1-like phenotype of HSALR mice, translation into the clinic would necessitate a delivery strategy that would expose multiple tissue types to the active morpholino oligonucleotide.
In the present work we tested a delivery strategy that would facilitate broad distribution of a CAG-based morpholino by employing recently described cationic peptides that enable systemic delivery of antisense cargo (Jearawiriyapaisarn et al., 2008). We set out to interrogate whether systemic delivery of peptide-morpholino conjugates tailored for DM1 would result in favorable biodistribution and allow the conjugates to reach a sufficient concentration in skeletal muscle to access the CUG repeat tract and mitigate RNA toxicity.
Materials and Methods
HSALR rederivation and genotyping
Rederived hemizygous HSALR mice were produced by impregnating FVB/n wild-type female mice with sperm from homozygous HSALR line 20b males. Male and female hemizygous mice were mated to generate homozygous offspring. A quantitative multiplex real-time polymerase chain reaction (RT-PCR) assay was used to determine the zygosity status of mice; this assay detects human skeletal actin (ACTA1) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh) as an internal control in the same well. The following primer-probe set (Life Technologies, Grand Island, NY) was used to detect ACTA1 genomic DNA: Forward: 5′-CCACCGCAAATGCTTCTAGAC; Reverse: 5′-CCCCCCCATTGAGAAGATTC; Probe: 5′-CTCCACCTCCAGCACGCGACTTCT. A proprietary sequence primer-probe set (Life Technologies) was used to detect Gapdh genomic DNA. Homozygous mice were subsequently back-crossed to wild-type FVB/n to confirm homozygosity status. Confirmed male and female homozygous mice were then mated to maintain a homozygous colony on an FVB/n background strain.
Conjugation of cell-penetrating peptides to morpholino oligonucleotides
Peptide-linked morpholino (PPMO) conjugates were synthesized as described by Abes et al. (Abes et al., 2006), with modifications. Peptide B (Ac(RXRRBR)2XB-OH) or peptide K (Ac(RXR)4XB-OH) (Anaspec, Fremont, CA) was activated in dimethylformamide containing O-(6-Chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU)/diisopropylethylamine (DIEA) at molar ratios of 1:2 per mole of peptide at room temperature (RT). Morpholino CAG25 (5′-AGCAGCAGCAGCAGCAGCAGCAGCA-3′) (Gene Tools, Philomath, OR) with a 5′ primary amine modification was dissolved in dimethylsulfoxide and added to activated peptide B or peptide K at a 1.2–1.5:1 molar ratio of peptide to ASO and the reaction was allowed to proceed for 2 hours at RT. The reaction was quenched with water; PPMO conjugates were purified over carboxymethyl sepharose (GE Healthcare, Piscataway, NJ) and eluted in 2M guanidine-HCl, 1M NaCl, pH 7.5, 20% acetonitrile. The eluate was dialyzed against several buffer exchanges of 0.1mM NH4HCO3 in a dialysis cassette with molecular weight cut-off of 3,000 Da. The dialyzed PPMO was quantified by spectrophotometric absorbance in 0.1N HCl at 265 nm, frozen, and lyophilized. Molecular weights of PPMO-B and PPMO-K were confirmed by matrix-assisted laser desorption/ionization mass spectrometry.
Intramuscular TA injections
The Genzyme Institutional Animal Care and Use Committee approved all animal studies. The TA of isoflurane-anesthetized mice was injected and subjected to electroporation as described (Wheeler et al., 2007). One TA was injected with 20 μg (1 μg/μL) of CAG25, GAC25 (5′-ACGACGACGACGACGACGACGACGA-3′) (Gene Tools) or PPMO (in PPMO-B and PPMO-K injections, 20 μg ASO equivalent mass was injected, taking into account the mass of the peptide), whereas the contralateral TA was injected with 20 μL phosphate-buffered saline (saline).
Systemic delivery studies in HSALR
CAG25, PPMO-B, and PPMO-K were dissolved in saline and administered to male and female HSALR homozygous mice at a dose of 30 mg/kg body weight by tail vein intravenous (IV) injection once a week for 6 weeks (saline, n=3–8; CAG25, n=6; PPMO-B, n=6; PPMO-K, n=3). Blood was collected by retro-orbital bleed 24 hours following the last dose for evaluation of serum chemistries. Approximately 1 week following the final dose, mice were evaluated for the presence of myotonia as described in electromyography procedures. Mice were then killed and muscle sections were frozen in liquid nitrogen for RNA analyses or embedded in optimal cutting temperature medium and frozen in cooled isopentane for immunofluorescence and fluorescence in situ hybridization (FISH) analyses.
RT-PCR analysis of alternative splicing
RT-PCR was carried out using the SuperScript 3 One-Step RT-PCR System with Platinum Taq DNA Polymerase (Life Technologies) with gene-specific primers used for complementary DNA synthesis and PCR amplification. The primer sequences for Serca-1, Zasp, Titin, and ClC-1 have been described previously (Lin et al., 2006; Wheeler et al., 2007). PCR products were electrophoresed on agarose gels, stained with SybrGreen 1 Nucleic Acid Gel Stain (Life Technologies), and imaged using a Fujifilm LAS-3000 Intelligent Dark Box.
Mbnl1 immunofluorescence microscopy
Frozen sections of quadriceps muscle 6 μm thick were processed to detect localization of Mbnl1 protein via immunofluorescence as previously described (Lin et al., 2006), with the following modifications: the rabbit polyclonal anti-Mbnl1 antibody A2764 was used at a concentration of 1:5,000, followed by incubation with Alexa Fluor 568-labeled goat-anti-rabbit secondary antibody at a concentration of 1:500. Samples were imaged using a LSM510 META laser scanning confocal microscope configured for imaging 4′,6-diamidino-2-phenylindole and Alexa Fluor 568 sequentially. Examination was conducted using a 100×/NA1.45 Plan-Fluor oil immersion objective with 4× zoom.
RT-PCR of HSALR transgene mRNA
Total RNA was purified from TA, gastrocnemius, and quadriceps using the RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Quantitative RT-PCR was used to determine the mRNA level of the HSALR transgene; 18S RNA level was used as a normalization factor. The primer-probe set used to detect ACTA1 mRNA has been described (Wheeler et al., 2012). The 18S RNA level was determined using a primer-probe set of proprietary sequences (Life Technologies).
Electromyography
EMG was performed upon isoflurane-anesthetized mice with 10 EMG samplings taken per muscle group in TA, gastrocnemius, and quadriceps using two 29-gauge needle electrodes. After insertion of both needles, the muscle was gently manipulated with forceps at the site of needle entry. Myotonia was graded via the following criteria: 0 indicates no myotonic discharges detected; 1 indicates less than 50% of muscle manipulations resulted in a myotonic discharge; 2 indicates 50% to 80% of muscle manipulations resulted in a myotonic discharge; and 3 indicates that 90% to 100% of muscle manipulations resulted in a myotonic discharge.
Results
Intramuscular injection of two different PPMO conjugates into TA muscle corrects aberrant RNA splicing
Jearawiriyapaisarn and colleagues have described several arginine-rich cell-penetrating peptide sequences that facilitate systemic biodistribution of morpholino ASOs when delivered as peptide conjugates via intravenous, intraperitoneal, or subcutaneous injection routes (Jearawiriyapaisarn et al., 2008). PPMO conjugates containing such cell-penetrating peptides, specifically peptide B and peptide K, have been studied extensively in mouse models of Duchenne muscular dystrophy. These studies demonstrated PPMO-mediated exon skipping of mutant dystrophin RNA in numerous muscle groups including TA, gastrocnemius, quadriceps, diaphragm, and heart (Fletcher et al., 2007; Wu et al., 2008; Goyenvalle et al., 2010). We reasoned that these cationic peptides may likewise enable systemic delivery of a CAG sequence-based morpholino to skeletal muscle, a key target tissue for a DM1 therapeutic. We therefore carried out condensation synthesis reactions to generate PPMO conjugates containing a 25-mer CAG sequence-based morpholino (CAG25; Wheeler et al., 2009) and a cell-penetrating peptide covalently appended to a spacer moiety located at the 5′ end of the morpholino (Fig. 1A). Two PPMO conjugates were synthesized containing either peptide B or peptide K and will be referred to as PPMO-B and PPMO-K respectively.
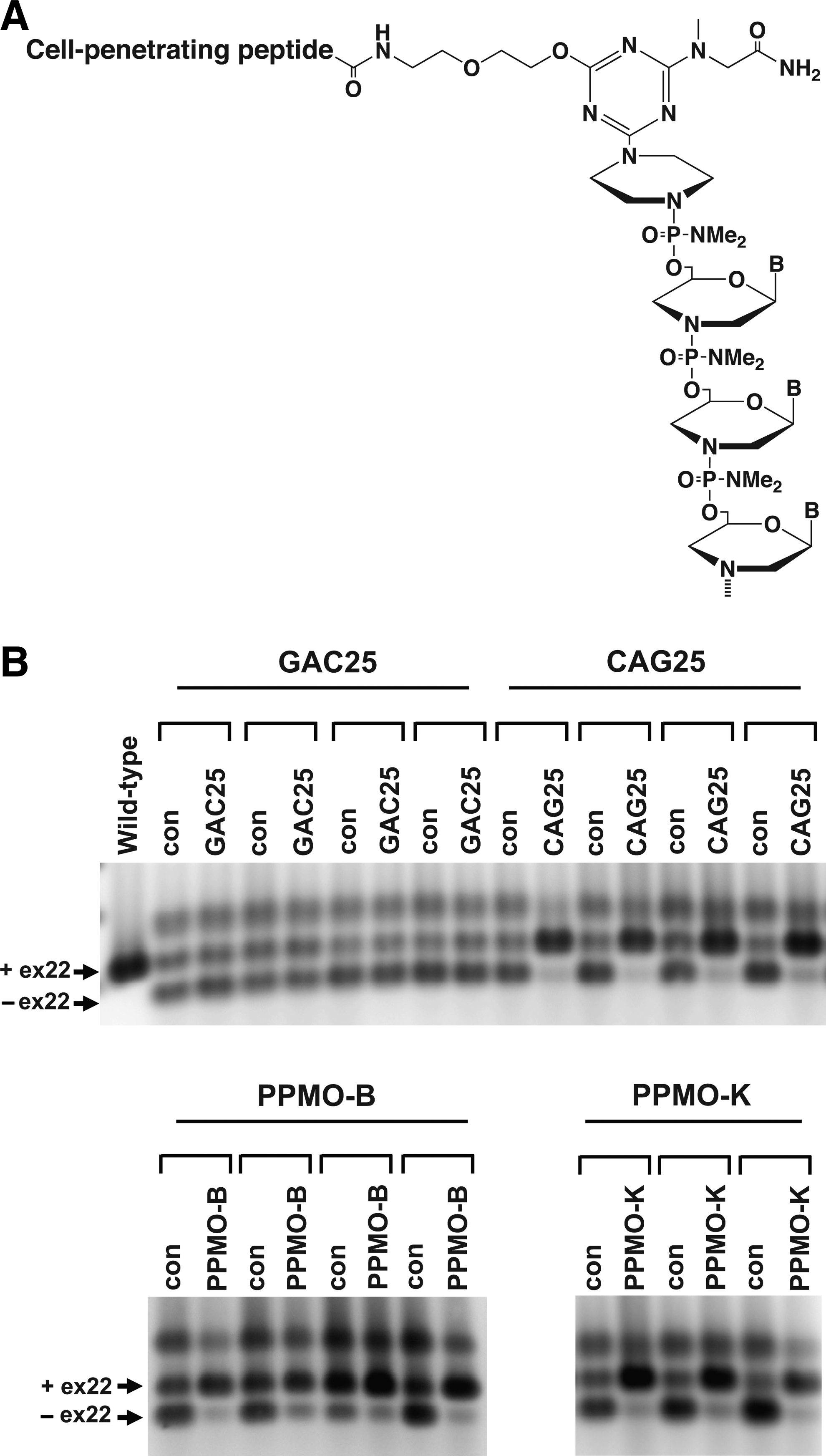
Chemical structure of peptide-linked morpholino (PPMO) and evaluation of the bioactivity of PPMO-B and PPMO-K in HSALR mice.
To determine the impact of the spacer and peptide modifications upon the bioactivity of PPMO-B and PPMO-K, we performed a series of IM injections into the TA muscle of HSALR transgenic mice and assessed RNA splicing of Serca-1; transcripts of Serca-1 exhibit altered RNA splicing in both HSALR and human DM1 patient skeletal muscle. As described by Wheeler et al., IM injection of CAG25 into the TA muscle of HSALR mice results in dispersal of RNA foci, relocalization of Mbnl1 protein into the nucleoplasm, and correction of Serca-1 splicing (Wheeler et al., 2009). As expected, IM injections of CAG25 into TA corrected Serca-1 splicing, whereas the contralateral TA that received an injection of saline retained the abnormal pattern of Serca-1 splicing (Fig. 1B). A control morpholino of GAC sequence (GAC25) did not correct Serca-1 splicing as anticipated. Importantly, IM injections of both PPMO-B and PPMO-K also corrected Serca-1 splicing indicating that the peptide and spacer covalent modifications at the 5′ end of the morpholino do not compromise the ability of CAG25 to interact with its target and neutralize the toxic effects of CUG RNA (Fig. 1B).
Repeated IV injections of PPMO-B and PPMO-K effectively ameliorate spliceopathy in vivo
Having confirmed that modification of CAG25 with peptide B or K did not abrogate its bioactivity, we proceeded to evaluate whether PPMO-B and PPMO-K could modulate the effects of toxic RNA in HSALR mice using an IV dosing regimen. Repetitive IV dosing had been demonstrated to restore dystrophin expression by PPMO conjugates designed to skip dystrophin exon 23 in the Mdx mouse model of Duchenne muscular dystrophy (Wu et al., 2008). We therefore subjected HSALR mice to 6 weekly IV injections of CAG25, PPMO-B, and PPMO-K at a dose of 30 mg/kg. HSALR mice tolerated this dose well and exhibited no overt signs of toxicity during CAG25 and PPMO administration or throughout the study period. Additionally, these treatments did not adversely affect levels of liver transaminases (ALT, AST) or markers of kidney function (creatinine, BUN) (Supplementary Fig. S1A, B; Supplementary Data are available online at www.liebertpub.com/nat). Additionally, no evidence of CAG25- or PPMO-related toxicity or accumulation was present histologically in the liver or kidney (Supplementary Fig. 1C, D).
We conducted splicing analyses on several muscle groups including the TA, quadriceps, and gastrocnemius. As shown in Fig. 2, both PPMO-B and PPMO-K dramatically corrected Serca-1 splicing and restored the pattern of splicing to that observed in wild-type mice. Correction of Serca-1 splicing occurred in all muscles examined including TA, quadriceps, and gastrocnemius. One mouse treated with PPMO-B showed partial splicing correction in quadriceps and gastrocnemius. In contrast to PPMO-B and PPMO-K, intravenous injections of unmodified CAG25 led to no detectable improvements in Serca-1 splicing in all muscle groups examined (Fig. 2). This is consistent with other studies showing inferior biodistribution of bare morpholinos to muscle in comparison to PPMO(Sazani et al., 2002; Jearawiriyapaisarn et al., 2008). Additionally, 6 weekly IV injections of PPMO-B at a lower dose of 10 mg/kg did not correct Serca-1 splicing (data not shown).
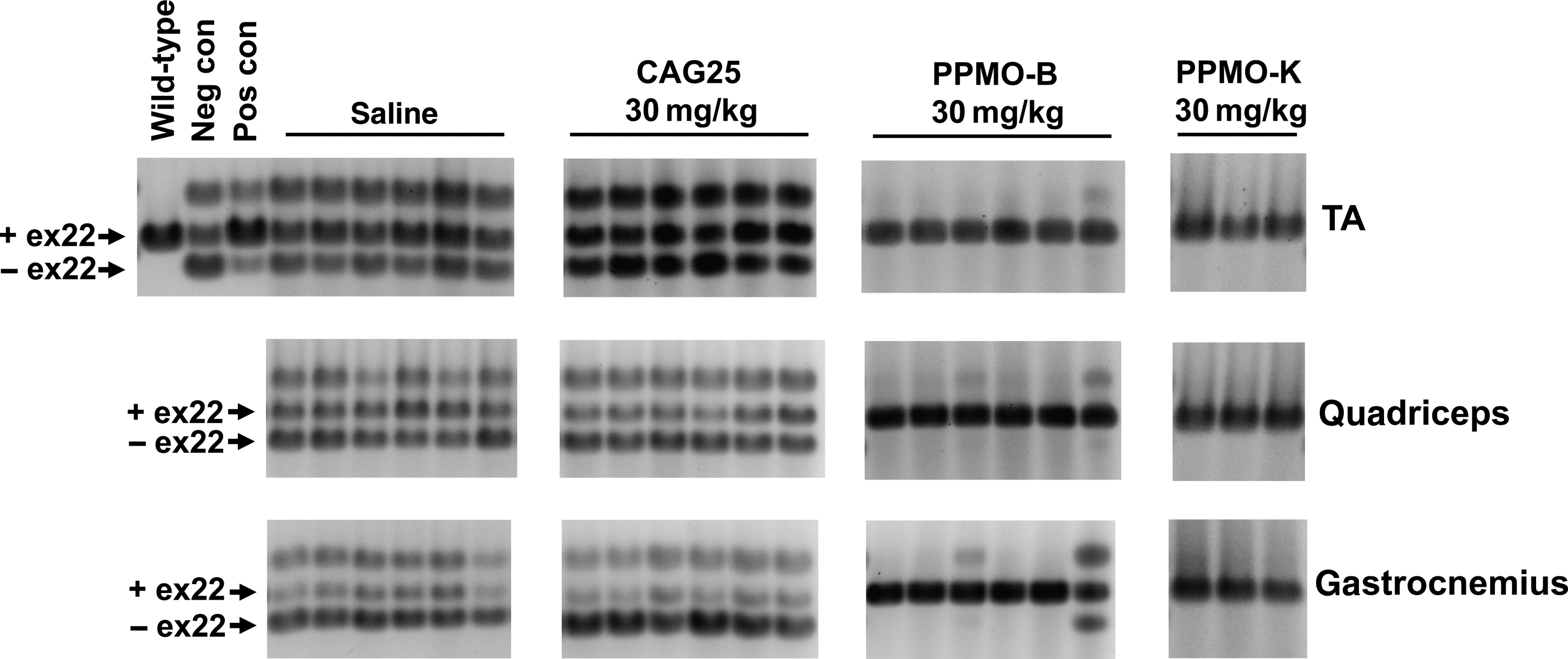
Repeated intravenous (IV) injections of PPMO-B or PPMO-K correct Serca-1 splicing. HSALR mice were injected with saline, CAG25, PPMO-B, or PPMO-K weekly for 6 weeks. The indicated muscle groups were collected from mice approximately 1 week following the last dose. Total RNA was purified, and Serca-1 was amplified to detect differentially spliced isoforms. RNA from the TA of a FVB/n wild-type mouse served as a control as in Fig.1. Serca-1 splicing in RNA purified from the TA of HSALR mice injected with GAC25 or CAG25 as in Fig. 1 served as a negative control (neg con) and a positive control (pos con), respectively.
Additional transcripts dependent upon MBNL1 for proper splicing in HSALR transgenic mice and human DM1 patients include Titin, Zasp, and ClC-1 (Mankodi et al., 2002; Lin et al., 2006). As shown in Fig. 3, systemic delivery of PPMO-K led to robust corrections in RNA splicing of Titin, Zasp, and ClC-1 in TA, quadriceps, and gastrocnemius. HSALR mice treated with IV injections of PPMO-B likewise demonstrated corrections in alternative splicing for these transcripts (data not shown). These results suggest that systemically delivered PPMO-B and PPMO-K were able to penetrate into skeletal muscle, enter into muscle myonuclei, hybridize with poly (CUG) RNA, and liberate an amount of sequestered Mbnl1 protein sufficient to restore correct Mbnl1-regulated RNA splicing.

Correction of abnormal splicing of ZASP, Titin, and ClC-1 in HSALR mice dosed with repeated IV injections of PPMO-K. HSALR mice were injected with saline or PPMO-K once a week for 6 weeks. The indicated muscle groups were collected from mice, total RNA was purified, and Zasp, Titin, and ClC-1 were amplified to detect differentially spliced isoforms containing the indicated exon (ex) and intron (int) configurations; for Titin, splice isoforms with m-line exon 5 inclusion (+m-ex5) or exclusion (– m-ex5) were amplified. RNA from the TA of a FVB/n wild-type mouse served as a non-disease control. Positive and negative controls were generated with RNA as in Fig. 2 with PCR amplification for Zasp, Titin, and ClC-1.
PPMO-K disrupts CUG nuclear inclusion complexes in skeletal muscle and results in a redistribution of Mbnl1 protein
To determine the fate of ribonuclear foci in mice treated with PPMO-K, we used FISH to detect CUG ribonuclear inclusions in muscle. In PPMO-K-treated HSALR mice, we observed a reduction in the number and intensity of CUG foci (Supplementary Fig. S2). However, because the oligonucleotide sequence of PPMO-K and the probe utilized in the FISH procedure consist of the same (CAG)n sequence, it is not possible to determine whether the effects upon RNA foci are due to disruption of the foci by PPMO-K or due to binding competition between PPMO-K and the FISH probe. As a more direct, unequivocal means to ascertain the effect of PPMO-K upon foci, we performed immunofluorescence labeling of Mbnl1, which is sequestered in CUG RNA foci and shows punctate staining in the nucleus of HSALR mice (Fig. 4A–C). As shown in Fig. 4D–F, administration of PPMO-K led to a redistribution of Mbnl1 protein and resulted in a more diffuse localization within the nucleoplasm that is similar to that noted in wild-type mice (Fig. 4G–I). Taken together, the immunostaining and FISH data suggest that PPMO-K interacts with CUG RNA in a competitive manner thereby leading to a redistribution of Mbnl1 protein within myonuclei. The liberation of Mbnl1 in skeletal muscle foci is consistent with the correction of RNA splicing observed in PPMO-K-treated mice.

Immunofluorescence microscopy demonstrates nuclear redistribution of muscleblind-like 1 (Mbnl1) protein in peptide-linked morpholino (PPMO)-K–treated mice. Immunofluorescence staining of Mbnl1 proteins was performed on sections of quadriceps muscle and confocal microscopy was carried out as described in the materials and methods.
Myotonia is corrected in HSALR mice subjected to IV injections of PPMO-K
The correction of ClC-1 RNA splicing by PPMO-B and PPMO-K (Fig. 3) suggests that restoration of chloride channel function may result in alterations in the severity of myotonia that is typically observed in HSALR mice. To test this possibility, we evaluated myotonia in saline-treated and PPMO-K-treated HSALR mice by EMG. We first assessed myotonia in wild-type FVB/n, hemizygous and homozygous HSALR mice. Hemizygous mice contain approximately 50% of the level of HSALR mRNA in skeletal muscle vs. homozygous mice (Fig. 5A). We detected a reduction in the occurrence of myotonia in hemizygous mice relative to homozygous mice and no myotonia in wild-type mice (Fig. 5C) as expected. A tracing of a typical myotonic discharge that we detected in homozygous HSALR mice is shown in Fig. 5B. As shown in Fig. 5C, no myotonia was detected in HSALR mice treated with PPMO-K, consistent with the corrections in ClC-1 alternative splicing that occurred in these mice.
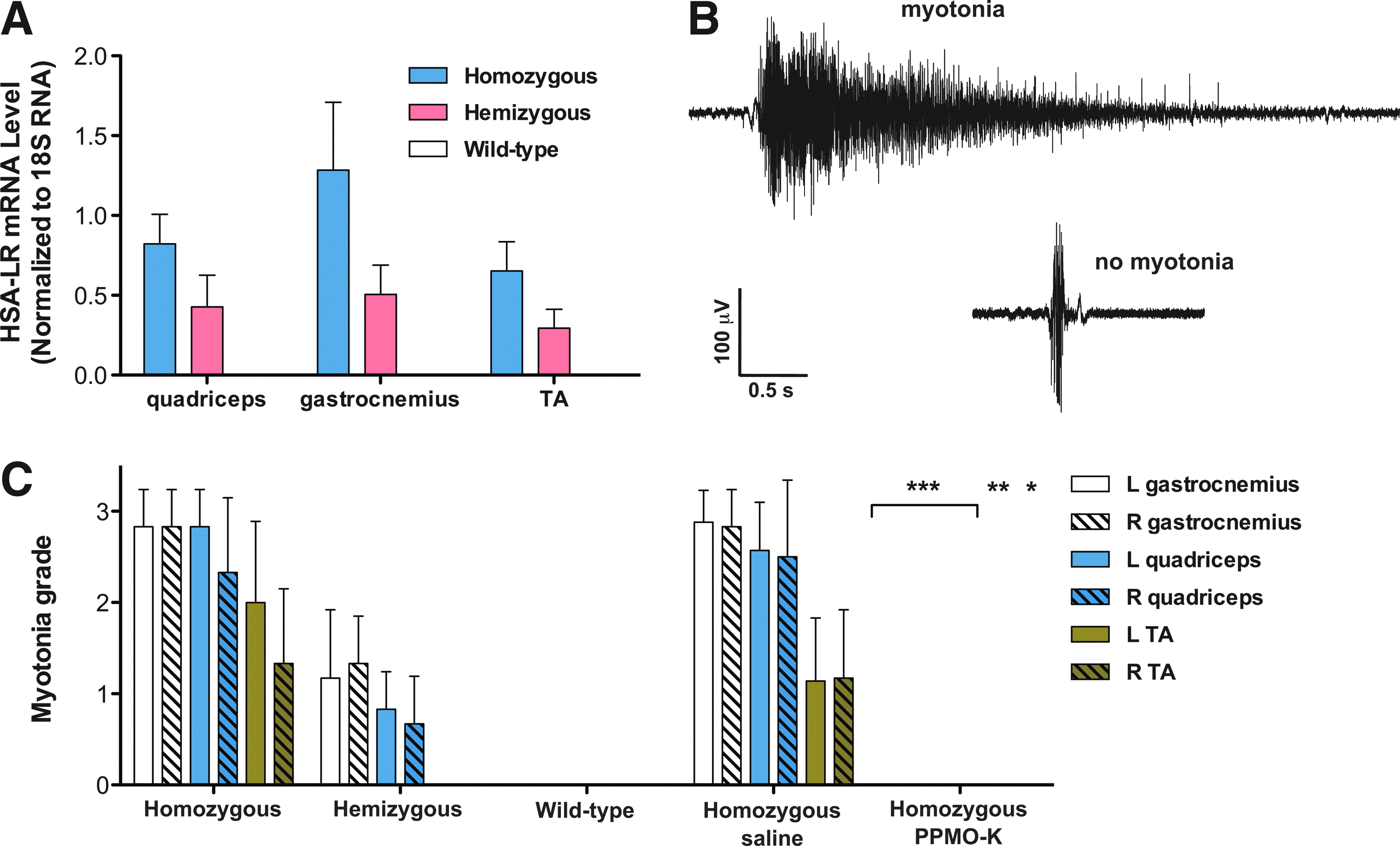
Myotonia is corrected in HSALR mice dosed with repeated IV injections of PPMO-K
Discussion
DM1 remains a disease with unmet medical need, and recent clinical trials addressing aspects of the disease such as myotonia and muscular atrophy have delivered mixed results. One clinical examination demonstrated that myotonia can be reduced with the sodium channel blocker mexiletine (Logigian et al., 2010), although this study assessed myotonia that was elicited by a voluntary handgrip; whether or not other muscle groups experience similar improvements with mexiletine treatment has yet to be determined. In an attempt to increase muscle mass and hence mitigate muscle atrophy and resulting functional deficits, recombinant human insulin-like growth factor 1 (rhIGF-1) in complex with recombinant human IGF binding protein 3 (rhIGFBP-3) (rhIGF-1/rhIGFBP-3) was evaluated in DM1 patients (Heatwole et al., 2011). Although increases in lean body mass were observed in DM1 subjects treated with rhIGF-1/rhIGFBP-3, measurements in quantitative functional and strength tests in muscle did not reveal significant improvements.
At the same time that these symptom-based interventions have been under examination in clinical trials, several initiatives have focused on developing molecules that bind to the central pathogenic mediator in DM1, the long repeats of CUG RNA. Several different classes of compounds that interact with nucleic acids have been described that bind to CUG RNA including small molecules, modular assemblies of CUG ligands, and ASOs containing various modifications (Kole et al., 2012) aimed at improving target affinity and in vivo stability. Polymeric assemblies of kanamycin (Lee et al., 2009) and Hoechst 33258 (Pushechnikov et al., 2009; Childs-Disney et al., 2012) were shown to bind CUG RNA and inhibit the CUG RNA–MBNL1 interaction. Likewise, a dynamic combinatorial selection method identified a series of disulfide-linked molecules that bound to CUG RNA and inhibited the interaction of MBNL1 with CUG RNA (Gareiss et al., 2008). Refinement of these molecules led to compounds that afforded partial correction of RNA splicing when delivered intraperitoneally to HSALR transgenic mice (Ofori et al., 2012). A chemical similarity search identified a compound similar to Hoechst 33258 that was able to displace MBNL1 from CUG RNA; this compound also partially corrected splicing in HSALR (Parkesh et al., 2012). The nucleic acid–binding molecule pentamidine was also shown to disrupt the MBNL1–CUG RNA interaction and provoked partial splicing correction in HSALR transgenic mice (Warf et al., 2009). The partial responses of these compounds in RNA splicing correction may indicate the need for improvements in the duration of exposure to target, target affinity, and/or biodistribution to muscle.
In addition to morpholino oligomers, 3 additional antisense chemistries have been utilized to target the CUG repeat tract in transgenic mouse models of DM1. Mulders and colleagues (Mulders et al., 2009) generated a 2′-O-methyl phosphorothioate–modified ASO of CAG sequence termed PS58 that preferentially led to reduction of mutant DMPK versus unaffected DMPK mRNA in myoblasts derived from DM1 patients. PS58 also reduced levels of HSALR mRNA by approximately 50% and generated minor corrections in RNA splicing when delivered directly to TA muscle of HSALR transgenic mice. In another effort targeting the CUG repeat, Lee and colleagues (Lee et al., 2012) used intramuscular injection of a 2′-MOE-modified, RNaseH-active ASO to invoke RNaseH-mediated degradation of transgene mRNA containing a CUG expansion in EpA960 transgenic mice; a 50% decrease in transgene mRNA was achieved with minor corrections in RNA splicing. More robust phenotype corrections were observed with systemically delivered 2′-MOE-modified, RNaseH-active ASOs that target HSALR transcripts for degradation via RNaseH (Wheeler et al., 2012). The stronger corrections in RNA splicing and the full elimination of myotonia observed in these studies (Wheeler et al., 2012) may be explained by the greater than 80% decrease in HSALR mRNA in hindlimb skeletal muscles.
In the present work we evaluated the approach of systemically delivering morpholino oligomers with cell-penetrating peptides in a widely used transgenic mouse model of DM1. We demonstrated that addition of a cell-penetrating peptide to a CAG sequence morpholino permitted sufficient uptake of PPMO into skeletal muscle to neutralize the toxic effects of an elongated CUG repeat. We observed near complete resolution of splicing defects, release of Mbnl1 from RNA foci, and elimination of myotonia. One consideration of the CUG neutralization strategy concerns the variability of CUG length that is present in individuals affected with DM1. In our studies, HSALR transgenic mice represent a patient population of similar CUG burden as determined by the 250 CTG trinucleotide repeat present in the HSALR transgene. CUG repeat length varies considerably among DM1 patients, bringing into consideration that the present treatment strategy, if tolerable and appropriate in human patients, could potentially require patient-specific tailoring of dose depending upon the amount of CUG mass present. ASOs that invoke RNaseH activity may overcome the need to titrate to an effective dose due to the catalytic nature of the mechanisms involved in RNaseH-mediated mRNA degradation. A second consideration of the present strategy concerns the findings of PPMO toxicity in kidney above a threshold dose in non-human primates, which may limit the therapeutic index for safe delivery to humans (Moulton and MOULTON, 2010; Kole et al., 2012).
In conclusion, the finding that systemic delivery of a morpholino oligonucleotide can modulate DM1-like pathology in vivo is multifold in significance: first, 5′ covalent modification of a CAG-based morpholino does not detract from interaction with target CUG RNA; second, the biodistribution that is rendered achievable by arginine-rich cell-penetrating peptides is sufficient to confer corrections in biochemical and physiological aspects of DM1 pathology; and thirdly, the test agent administered in the studies presented here in the HSALR model of DM1 is the same therapeutic candidate that could be evaluated for safe use in human DM1 patients.
Footnotes
Acknowledgments
We thank Thurman Wheeler and Charles Thornton for guidance in setting up the electroporation procedure in TA muscle and for kindly providing Mbnl1 polyclonal antibody A2764. We are grateful to Emily Pease and Stan Nakanishi for assistance in setting up electromyography equipment and procedures. We thank Joanne Cotton for performing mass spectrometry analysis on PPMO-B and PPMO-K. We thank and acknowledge the Genzyme histology and pathology departments. We thank Michelle Searles for performing serum chemistry measurements. We also thank Patrick DeCourcy, Matt DeRiso, and Mike Phipps for genotyping HSALR mice and Greg Ulinski for assistance with confocal microscopy. We are grateful to Leah Curtin, Biomedical Research Models staff, and Charles River Laboratory staff for HSALR colony management. We thank Ronald Scheule and Nelson Yew for critically reviewing the manuscript.
Author Disclosure Statement
A.J.L., L.M.M., N.P.C., I.W., T.W., L.P., E.R., P.A.P., S.H.C., and B.M.W. are employees of Genzyme, a Sanofi Company.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.