
Abstract
Small interfering RNAs (siRNAs) silence gene expression by triggering the sequence-specific degradation of mRNAs, but the targeted delivery of such reagents remains challenging and a significant obstacle to therapeutic applications. One promising approach is the use of RNA aptamers that bind tumor-associated antigens to achieve the delivery of siRNAs to tumor cells displaying specific antigens. Wholly RNA-based constructs are advantageous because they are inexpensive to synthesize and their immunogenicity is low. We therefore joined an aptamer-recognizing alpha V and integrin beta 3 (αvβ3) integrin to a siRNA that targets eukaryotic elongation factor 2 and achieved for the first time the targeted delivery of a siRNA to tumor cells expressing αvβ3 integrin, causing the inhibition of cell proliferation and the induction of apoptosis specifically in tumor cells. The impact of our results on the development of therapeutic aptamer–siRNA constructs is discussed.
Introduction
In the last 20 years, several aptamers have been identified as promising pharmaceutical candidates and have been tested as therapeutics because of their antiviral (Fukuda et al., 2000; Proske et al., 2002), anti-angiogenesis (Ng et al., 2006), anti-inflammatory (Jeong et al., 2001; Pietras et al., 2001), anticoagulation (Rusconi et al., 2002) and anti-proliferation characteristics (Mi et al., 2005; Li et al., 2011).
Aptamers have several distinctive properties that make them potentially useful therapeutic agents compared to full size or single chain antibodies. First, aptamers can be designed and chemically selected in vitro to bind any protein (including toxins and non-immunogenic targets). Second, the chemical synthesis of aptamers means there is less batch-to-batch variation. Third, there is no evidence that aptamers induce an immune response. Fourth, aptamers can be chemically modified to generate derivatives with diverse properties. Finally, the shelf life of aptamers is unlimited and they can be transported at room temperature.
Different aptamers that bind a wide range of cell-surface receptors have been selected and used for drug delivery by receptor-mediated endocytosis. In this context, aptamers can be used to deliver chemotherapeutic agents (Farokhzad et al., 2006; Dhar et al., 2008; Huang et al., 2009), photosensitizers (Mallikaratchy et al., 2008; Ferreira et al., 2009) and gold particles (Elbakry et al., 2009).
We have previously demonstrated the therapeutic potential of siRNA transcripts targeting eukaryotic elongation factor 2 (EEF2), which induce apoptosis in target cells (Wullner et al., 2008). We joined the siRNA to an RNA aptamer recognizing prostate-specific membrane antigen (PSMA) and achieved the targeted destruction of prostate cancer cells. We reasoned that aptamers targeting different tumor-specific receptors should achieve similar results in other cancer cell populations and chose integrin alpha V and integrin beta 3 (αvβ3) as the next target.
Integrins are heterodimeric transmembrane glycoproteins that mediate cell adhesion and play an important role in the regulation of tissue integrity, tissue remodeling, cell proliferation, cell migration, differentiation, apoptosis, vascular healing, and tumor invasion. The αvβ3 integrin binds vitronectin and is strongly expressed in certain tumor cells (Luo et al., 2007; Yoshimoto et al., 2008; Taherian et al., 2011) including prostate tumors (Goel et al., 2008), cervical tumors (Chattopadhyay and Chatterjee, 2001) and brain tumors (Takano et al., 2000). We achieved the targeted delivery of anti-EEF2 siRNA (siEEF2) to human glioblastoma, cervical cancer, and prostate cancer cells using aptamers that bind specifically to αvβ3 integrin (Mi et al., 2005), inhibiting cell proliferation, and triggering apoptosis specifically in the target cell population.
Materials and Methods
Preparation of siRNAs
Synthetic 21-nt RNAs were purchased from Qiagen (Hilden). The siEEF2 sequences were 5′-GCG CCA UCA UGG ACA AGA AUU dTdT (sense) and 5′-UUC UUG UCC AUG AUG GCG CGG dGdG (antisense). The negative control non-silencing siRNA (siNON) sequences were 5′-UUC UCC GAA CGU GUC ACG UdTdT (sense) and 5′-ACG UGA CAC GUU CGG AGA AdTdT (antisense). TOX™ transfection control siRNA (Dharmacon) was used as a positive control for transfection efficiency and toxicity. Transfections were carried out using HiPerFect transfection reagent (Qiagen, Hilden) according to the manufacturer's protocol.
Cell lines
We used 3 cell lines that expressed αvβ3 integrin strongly. The human glioblastoma cell line U-87 MG (ATCC No. HTB-14™) was grown in Dulbecco's Modified Eagle Medium (DMEM) containing 1 g/L glucose and 1% MEM non-essential amino acids. The human cervical carcinoma cell line SiHa (ATCC No. HTB-35) was grown in DMEM supplemented with 4.5 g/L glucose. The human prostate carcinoma cell line PC-3 (ATCC No. CRL-1435™) was grown in RPMI-1640 medium. As a control, we used the human embryonic kidney cell line HEK-293T (ATCC No. CRL-11268) which expresses only low levels of αvβ3 integrin and this was also grown in RPMI-1640 medium. All media were supplemented with 10% heat-inactivated fetal bovine serum and 100 U/mL penicillin streptomycin. The cells were incubated at 37°C under a 5% CO2 atmosphere. All cell culture media and additives were sourced from GIBCO® Cell Culture/Invitrogen.
Preparation of aptamer-siRNA chimeras
Predicting RNA secondary structure
The first step in preparation of aptamer–siRNA chimeras was to predict the secondary structure of the anti-αvβ3 integrin aptamer (Apt-αvβ3) (Mi et al., 2005), the aptamer–siRNA construct (Apt-αvβ3–siEEF2), and the control construct Apt-αvβ3–siNON (Table 1). All RNA secondary structures were predicted using the MFOLD server (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form), and the most stable structures with the lowest free energies for each construct were compared.
αvβ3, alpha V and integrin beta 3; EEF2, eukaryotic elongation factor; siRNA, small interfering RNA.
DNA template construction
Apt-αvβ3, Apt-αvβ3-siEEF2, and Apt-αvβ3-siNON template DNAs for in vitro transcription were designed, incorporating the T7 RNA polymerase promoter using Assembly PCR Oligo Maker (Rydzanicz et al., 2005) (http://startrek.ccs.yorku.ca/∼pjohnson/AssemblyPCRoligomaker.html). The templates were synthesized by assembly-polymerase chain reaction (PCR) using 6 overlapping primers (Table 2). Each reaction mixture contained 2.5 μM of each primer, 0.2 mM of each dNTP, and 2.5 U of GoTaq DNA polymerase in a final volume of 100 μL, and was amplified at 94°C for 10 minutes followed by 30 cycles of 94°C for 1 minute, 55°C for 1 minute, and 72°C for 1 minute, with a final incubation at 72°C for 10 minutes.
In vitro transcription
Apt-αvβ3-siEEF2 and Apt-αvβ3-siNON transcripts were synthesized in vitro in a 20-μL reaction using the amplified dsDNA template is described above and the DuraScribe T7 Transcription Kit (Epicentre). The reaction comprised 1 μg DNA template, 5 mM each of ATP and GTP, 5 mM each of 2′F–dCTP and 2′F-dUTP, and 2 μL DuraScribe T7 enzyme mix. Reactions were carried out for 6 hours at 37°C followed by DNase1 treatment for 20 minutes at 37°C. The RNA products were purified by denaturing (7 M urea) 8% polyacrylamide gel electrophoresis. RNA bands were excised and RNA was eluted in 0.3 M sodium acetate for 1 hour at 60°C and recovered by ethanol precipitation.
Fluorescence labeling of RNA
The 5′-ends of the substrate RNA strands were modified during in vitro transcription using 25 mM guanosine-5′-O-monophosphothioate (GMPS), 2.5 mM of each NTP, and 2 μL DuraScribe T7 enzyme mix. The in vitro transcript was digested with DNase1 for 20 minutes, and the GMPS thiol group was reduced with 100 mM dithiothreitol (DTT). After purifying the RNA, the reduced thiol group was labeled with the fluorescent probe 5′-iodoacetamidofluoresceine (IAF) in labeling buffer [10 mM ethylenediaminetetraacetic acid (EDTA), 1 M urea, 100 mM Tris-HCl, pH 7.4] with 10% (v/v) dimethylformamide overnight at 4°C.
Labeled transcripts were purified by high-performance liquid chromatography (Shimadzu Prominence HPLC system) and monitored by absorption spectroscopy at 260 and 480 nm. We used a Water XBridge™ OSTC18, 2.5μm (4.6×50 mm) column (Waters) for RNA analysis and purification. The mobile phase flow rate was 1 mL/min. During the analytical and purification runs, gradient elution was carried out using mobile phases A [0.1 M triethylammonium acetate (TEAA) and 5% acetonitrile)] and B (0.1 M TEAA and 30% acetonitrile). The RNA was separated using a 20-minute gradient of B from 0 to 100%.
Flow cytometry
Apt-αvβ3–siEEF2 RNA was refolded by heating for 5 minutes at 80°C, then slowly cooled to 37°C and incubated at 37°C for 10 minutes in 1×4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (20 mM HEPES, 150 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, pH 7.4). Approximately 4×105 U-87 MG, SiHa, PC-3, and HEK-293T cells were incubated with 200 nM of labeled RNA in 1× HEPES buffer or with the phycoerthrin (PE)-labeled monoclonal anti-human αvβ3 integrin antibody (R&D Systems GmbH) for 30 minutes on ice. The cells were then washed twice with 1.8 mL 1×HEPES buffer and analyzed using a FACSCalibur device (Becton & Dickinson). The resulting data were analyzed with CellQuest Software (Beckton Dickenson).
Internalization assay
Approximately 4×105 U-87 MG, SiHa, PC-3, and HEK-293T cells were washed twice with 1.8 mL 1×HEPES buffer and incubated with 200 nM IAF-labeled RNA in 1×HEPES binding buffer supplemented with 16 mM glucose. The “no internalization” controls were kept on ice. All other samples were incubated in the dark at 37°C for 60 minutes. At the end of the incubation period, the cells were washed twice, as above, and the cell suspension was incubated for 5 minutes with 2 μL of a 1:100 diluted Draq5 solution (Biostatus) to counterstain the nucleus. All images were captured using a TCS SP5 confocal microscope (LEICA Microsystem).
Cytotoxic activity of Apt-αvβ3–siEEF2
Cell viability was determined using a sodium 2,3,-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) cell proliferation kit based on 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (Roche). Briefly, 2×104 U-87 MG, SiHa, PC-3, and HEK-293T cells were seeded in 96-well plates and incubated overnight at 37°C. The cells were treated with different concentrations (0, 100, 250, 500, 1,000, and 2,000 nM) of Apt-αvβ3–siEEF2 or Apt-αvβ3–siNON followed by incubation for 48 hours at 37°C, whereas 5 μg/mL zeocin was used as a positive control. The same cells were transfected with siEEF2 or siNON using HiPerFect transfection reagent (Qiagen) according to the manufacturer's protocol and incubated under the same conditions. After incubation, cells were treated for 4 hours with 1 mg/mL XTT solution at 37°C. Cell viability was determined by monitoring of the reduction of XTT to formazan at an absorbance wavelength of 450 nm and a reference wavelength of 630 nm using an ELx808 microtiter plate reader (BioTek Instruments GmbH).
The induction of apoptosis in U-87 MG, SiHa, PC-3, and HEK-293T cells was measured using the Apo-ONE Caspase-3/7 assay (Promega) according to the manufacturer's instructions. Briefly, cells were seeded in 96-well plates at a density of 2×104 cells per well in 100 μL medium and allowed to attach overnight at 37°C. Different concentrations (0, 100, 250, 500, 1,000, and 2,000 nM) of siEEF2, siNON, Apt-αvβ3-siEEF2, and Apt-αvβ3-siNON were added to the cells, whereas 5 μg/L zeocin was used as a positive control. HiPerFect transfection reagent was used to transfect the same cells with siEEF2 or siNON, followed by incubation under the conditions described above. After 48 hours of incubation at 37°C, 100 μL of Apo-ONE reagent containing a fluorogenic substrate was added to the cells. After 6 hours of incubation at room temperature, the proteolytic activity of caspase 3/7 was measured fluorimetrically (excitation wavelength 485 nm, emission wavelength 535 nm) using a microtiter plate reader.
Interferon assay
The induction of interferon β was measured using the human interferon β Enzyme-linked immunosorbent assay (ELISA) kit (PBL, Biomedical Laboratories) following the manufacturer's recommendations. Briefly, U-87 MG, SiHa, and PC-3 cells were incubated for 48 hours at 37°C after exposure to 2 μM Apt-αvβ3-siEEF2 or Apt-αvβ3-siNON. The cell culture supernatant (100 μL) was then transferred to a pre-coated ELISA plate and incubated for 24 hours at room temperature. Bound interferon β was detected by specific antibodies provided with the ELISA kit. Different concentrations (25, 50, 500, 1,000, and 2,000 pg/mL) of human interferon β were used as positive controls (provided in the kit).
Data analysis
Statistical analysis and curve fitting were carried out with GraphPad Prism software (GraphPad). Data represent the average of triplicates±standard error of the mean. Student's t-test and 2-way analyses of variance were used to assess the significance of independent experiments, with P<0.05 as the threshold for statistical significance.
Results
Design of the aptamer–siRNA construct
The secondary structure of Apt-αvβ3 RNA (the aptamer binding specifically to integrin αvβ3) was predicted using the MFOLD web server (ZUKER, 2003). As shown in Fig. 1, adding the siRNA sequence to Apt-αvβ3 RNA did not affect its predicted secondary structure.

MFold-predicted RNA secondary structures for the aptamer and aptamer–siRNA transcripts.
Binding of aptamer–siRNA transcripts to the cell surface
The expression of αvβ3 integrin on the surface of U-87 MG, SiHa, PC-3, and HEK-293T cells was confirmed by staining with a commercially available monoclonal anti-human αvβ3 integrin antibody. U-87 MG cells express abundant αvβ3 integrin and showed the highest binding FL-2 shift, whereas SiHa and PC-3 cells express moderate levels and showed smaller shifts, and HEK-293T cells express low levels and showed a minimal shift (Fig. 2B).
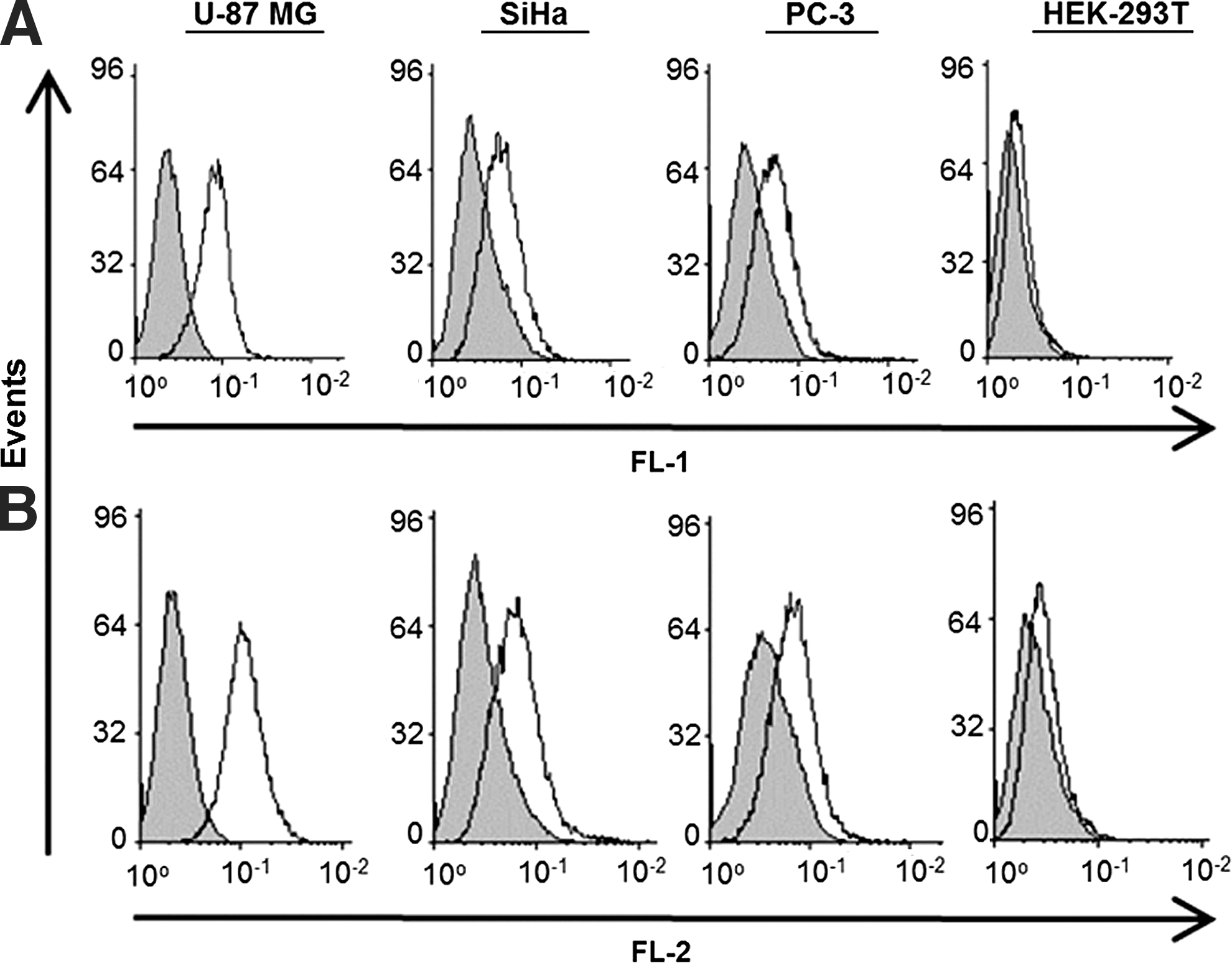
Binding activity of Apt-αvβ3 toward different antigen-positive tumor cell lines. Flow cytometry analysis was performed after incubating 4 × 105 cells with fluorophore-conjugated 5'-iodoacetamidofluoresceine (5-IAF) RNA or phycoerthrin (PE) αvβ3-specific positive control antibody for 30 minutes at 4°C in binding buffer.
The binding efficiency of the aptamer–siRNA transcripts was analyzed using the FL-1 channel of the flow cytometer. All 4 cell types were incubated with Apt-αvβ3-siEEF2 conjugated to 5-iodoacetamidofluorescein (IAF), and we observed efficient binding to U-87 MG, SiHa, and PC-3 cells, but not to HEK-293T cells (Fig. 2A).
Aptamer–siRNA internalization
All 4 cell types were incubated with IAF-labeled Apt-αvβ3-siEEF2 at 4°C or 37°C for 1 hour, and the internalization of the chimeric RNA was investigated by confocal microscopy. This revealed strong, specific, and homogeneous membrane staining on U-87 MG cells, moderate staining on SiHa cells, weaker but still evident staining on PC-3 cells, and no signal was detected on HEK-293T cells at 4°C (Fig. 3A), whereas at 37°C we also observed comparable levels of internal staining indicating the transcript had been taken up into U-87 MG, SiHa and PC-3 cells but not HEK-293T cells (Fig. 3B).
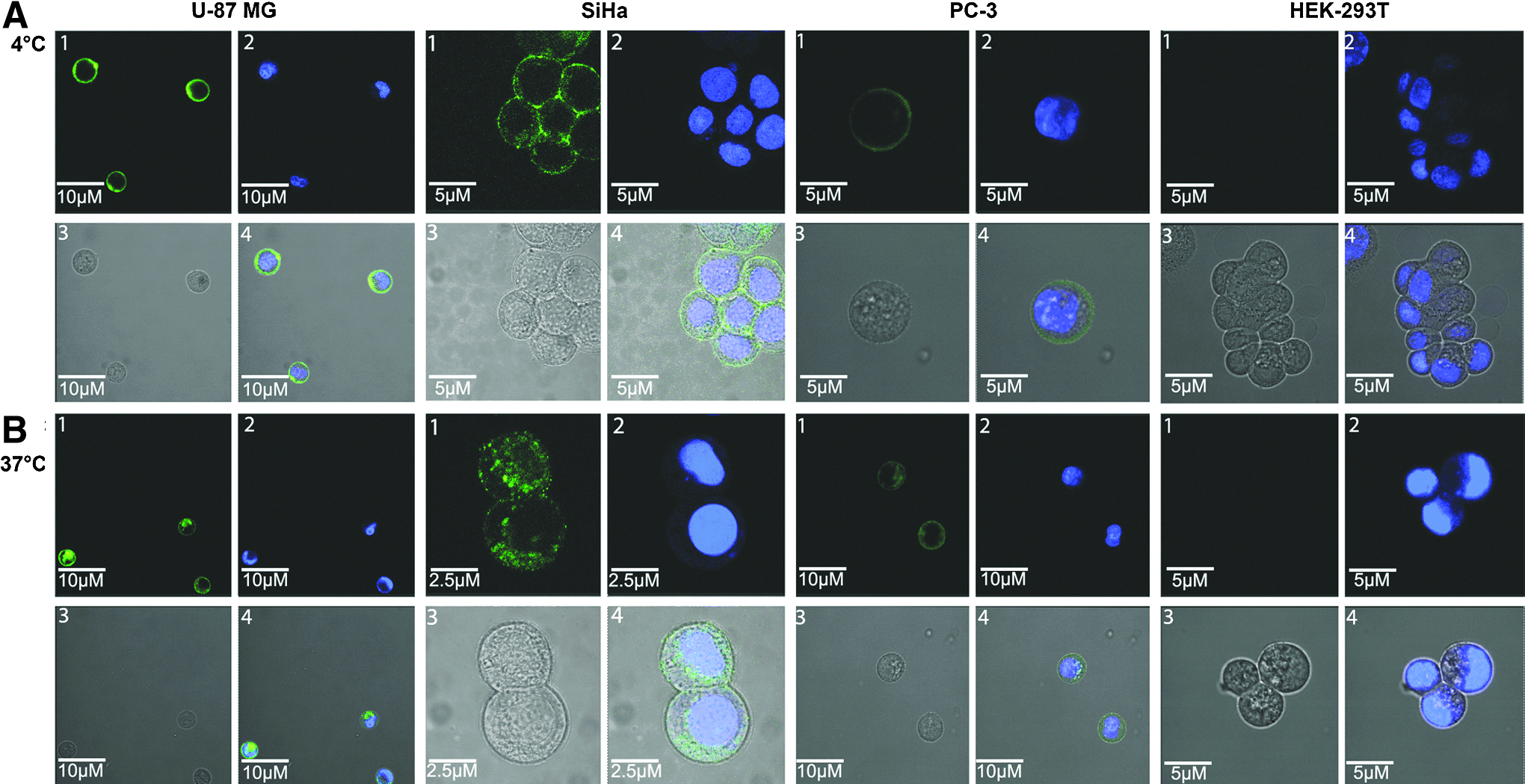
Confocal images of U-87 MG, SiHa, PC-3, and HEK-293T cells. IAF-Apt-αvβ3-siEEF2 was incubated with U-87 MG, SiHa, PC-3, and HEK-293T cells for 60 min at
Aptamer–siRNA-mediated cytotoxicity
The cytotoxic effects of Apt-αvβ3-siEEF2 and siEEF2 RNA against all 4 cell types were evaluated using an XTT-based colorimetric cell proliferation assay, with siNON and Apt-αvβ3–siNON as negative controls (Fig. 4). The viability of U-87 MG, SiHa, and PC-3 cells exposed to Apt-αvβ3-siEEF2 and siEEF2 for 48 hours was reduced significantly, in a concentration-dependent manner. The IC50 values for siEEF2 were 457 nM (U-87 MG), 549 nM (SiHa), 660 nM (PC-3), and 691 nM (HEK-293T), whereas those for Apt-αvβ3-siEEF2 were 708 nM (U-87 MG), 1,621 nM (SiHa), and 1,513 nM (PC-3). HEK-293T cells remained unaffected even when treated with 2,000 nM Apt-αvβ3-siEEF2. No significant toxicity was observed after treating the cells with siNON and Apt-αvβ3-siNON.
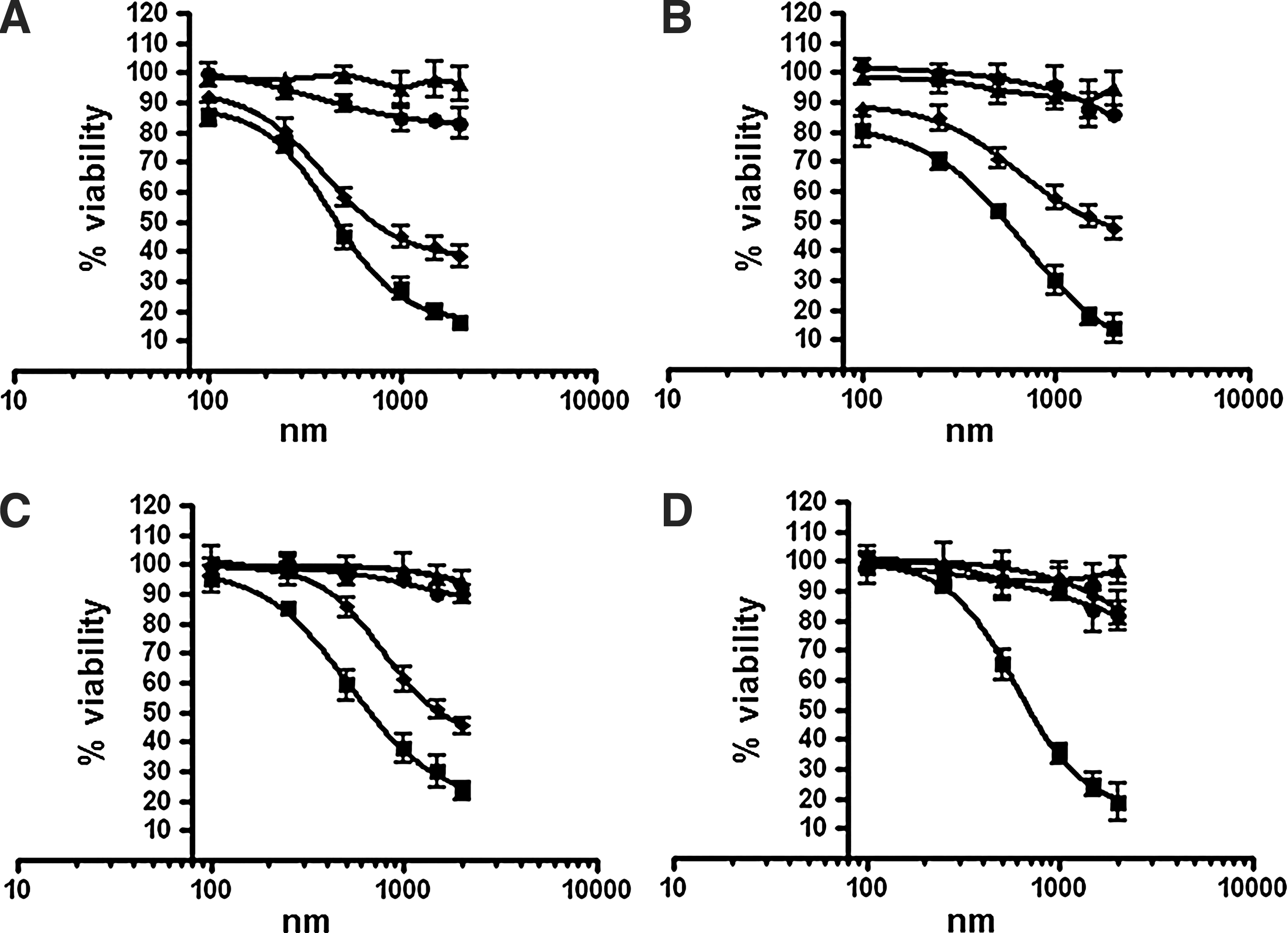
Cell viability analysis of Apt-αvβ3-siRNA. The cytotoxicity of siEEF2 (■) and Apt-αvβ3-siEEF2 (♦) was evaluated against
Aptamer–siRNA induced apoptosis
To determine whether Apt-αvβ3-siEEF2 could induce apoptosis in target cells, we measured the activation of caspase-3/7 in all 4 cell types after 48-hour exposure to different concentrations of the siRNAs and aptamer–siRNAs discussed above. We found that siEEF2 increased caspase-3/7 enzymatic activity in U-87 MG, SiHa, PC-3, and HEK-293T cells, whereas Apt-αvβ3siEEF2 affected U-87 MG, SiHa, and PC-3 cells but not HEK-293T cells (Fig. 5). Apoptosis was not induced in cells treated with siNON or Apt-αvβ3-siNON.
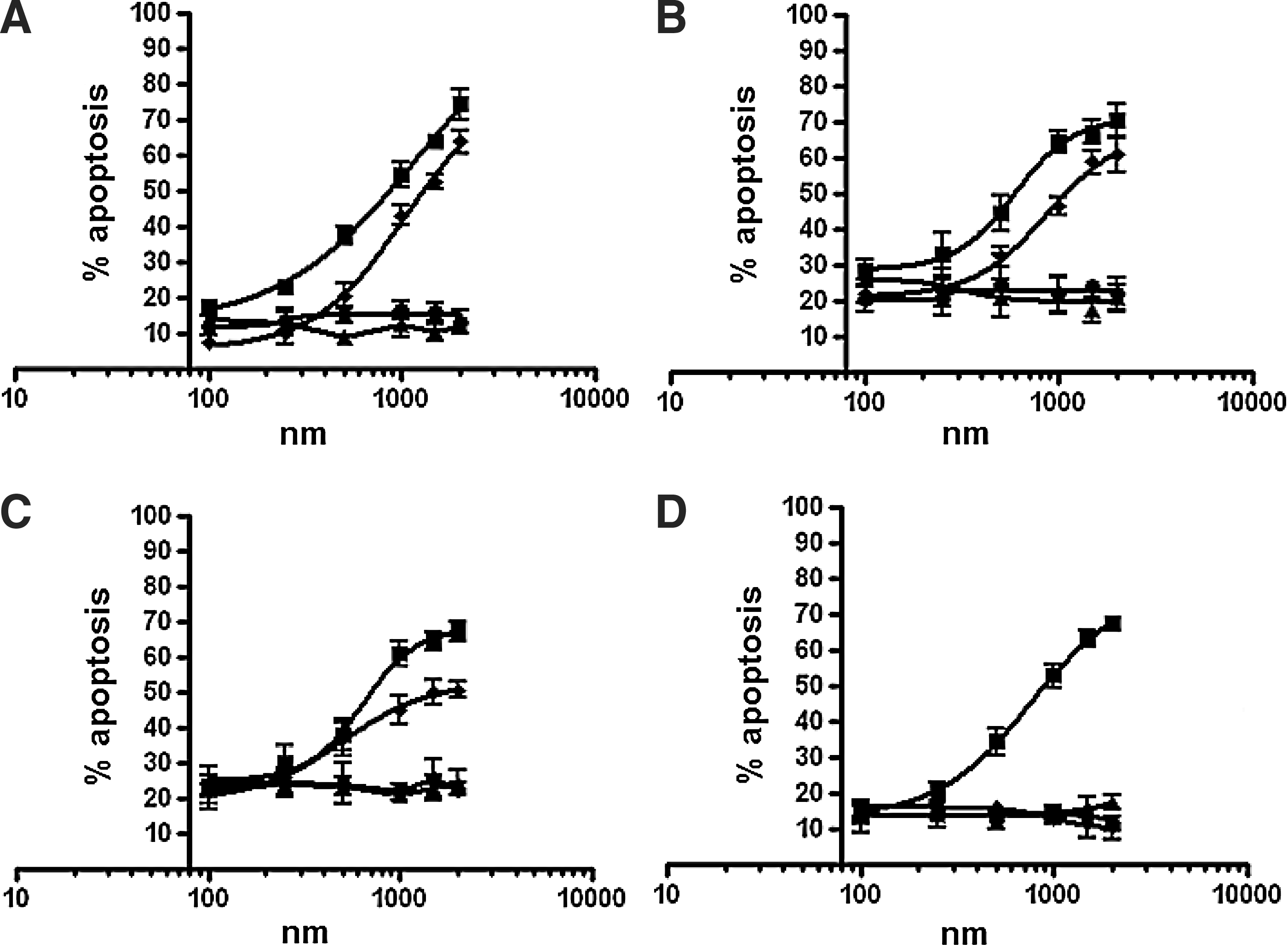
Induction of apoptosis by siEEF2 (■) and Apt-αvβ3–siEEF2 (♦) in
Interferon assay
One of the most important advantages of RNA-based drugs is their low immunogenicity compared with antibodies and other proteins. We investigated the immunogenicity of our construct by analyzing the production of interferon β after treating U-87 MG, SiHa, and PC-3 cells with the aptamer–siRNA transcripts. We found that treatment did not increase interferon β levels above those observed in untreated cells, indicating that under our experimental conditions the aptamer–siRNA transcripts did not trigger a type-1 interferon response (Fig. 6).
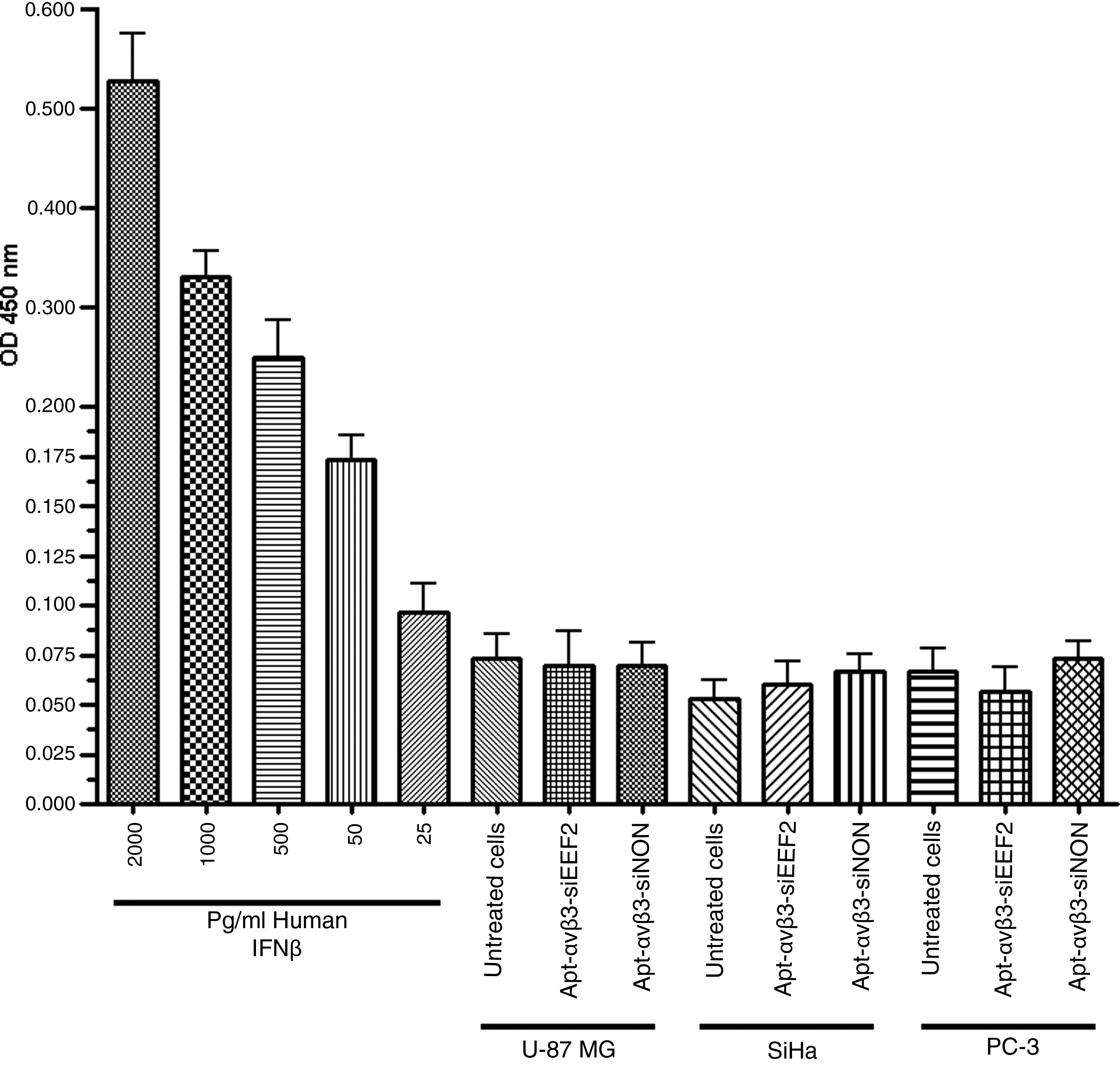
Interferon β detection assay. U-87 MG, SiHa, and PC-3 cells were incubated for 48 hours with 2 μM of RNA transcripts, and then the secretion of interferon β into the cell culture supernatant was analyzed using an interferon β sandwich ELISA kit. Different concentrations of human interferon β (hIFN-β) were used as a positive control, as shown in the left panel (filled gray bars).
Discussion
The potent ability of small interfering RNAs (siRNAs) to degrade specific target messenger RNAs and therefore inhibit gene expression makes therapeutic siRNAs promising new candidates for the treatment of cancer (Nguyen et al., 2008; Jeong et al., 2009; Leng et al., 2009). However, one of the greatest challenges facing RNA interference–based cancer therapy and preventing a breakthrough into the clinic is the inefficient delivery of siRNA across the plasma membrane of target cells. Among several approaches developed to address this issue, the use of aptamers as a targeting moiety is the most practical because the immunogenicity of aptamer-siRNA transcripts is low and they can be synthesized easily and inexpensively (Chu et al., 2006; McNamara et al., 2006; Wullner et al., 2008; Zhou et al., 2008; Neff et al., 2011).
The delivery of siRNAs to tumor cells has been achieved using aptamers, but thus far all reports have described the use of PSMA-specific aptamers that target prostate cancer cells. Many integrins are strongly expressed in tumor cells (Guo and Giancotti, 2004; Luo et al., 2007; Yoshimoto et al., 2008; Taherian et al., 2011), including αvβ3 integrin, which is expressed in several types of tumor cells (Takano et al., 2000; Chattopadhyay and Chatterjee, 2001; Goel et al., 2008). Several studies have demonstrated that integrins are internalized into endosomal compartments and then recycled back to the plasma membrane before eventual degradation (BRETSCHER, 1992; Caswell and Norman, 2006; White et al., 2007). The αvβ3 integrin is also recycled rapidly, returning to the cell surface within 30 minutes (Roberts et al., 2004; Woods et al., 2004; White et al., 2007). The strong expression of αvβ3 integrin in cancer cells and the efficient and rapid recycling via endocytosis are beneficial for the delivery of therapeutic agents.
We used Apt-αvβ3, an aptamer recognizing αvβ3 integrin, to deliver siEEF2, a siRNA that targets eukaryotic elongation factor 2 (Wullner et al., 2008), to U-87 MG, SiHa, and PC-3 cells, representing brain, cervical, and prostate cancers. Apt-αvβ3 has been shown to bind specifically to αvβ3 integrin expressed on the surface of human umbilical vein endothelial cells (Mi et al., 2005). Analysis of the secondary structure of the aptamer–siRNA chimera confirmed that the siRNA does not affect the folding of the aptamer. This was also confirmed by flow cytometry, which showed that Apt-αvβ3–siEEF2 is sufficient to target αvβ3 integrin on the surface of all 3 tumor cell types expressing this molecule, but not HEK 293T cells in which this module is expressed at minimal levels. Similar results were obtained using an anti-PSMA RNA aptamer linked to a siRNA sequence against elongation factor 2 gene, without effecting the secondary structure of anti-PSMA aptamer (Wullner et al., 2008).
Confocal microscopy revealed strong, specific, and homogeneous membrane staining when U-87 MG, SiHa, and PC-3 cells were incubated with IAF-labeled Apt-αvβ3-siEEF2. After 1 hour at 37°C, the IAF-Apt-αvβ3-siEEF2 was efficiently taken up into all 3 tumor cell lines (but not into HEK-293T cells) whereas the chimeric RNA was not taken up into any of the 4 cell types after 1 hour at 4°C. This confirmed that the uptake of Apt-αvβ3-siEEF2 is an active process involving receptor-mediated endocytosis.
EEF2 controls mRNA translation by translocating of the aminoacyl tRNA from the A site to the P site of the ribosome during polypeptide elongation (Boyce et al., 2008; Kaul et al., 2011). Therefore, targeted delivery of siRNA is critical to avoid the destruction of healthy cells. The αvβ3 integrin is overexpressed on angiogenic endothelial cells but not on normal cells, and the molecule is internalized when activated by its ligand (Chen and CHEN, 2011). These properties make αvβ3 integrin a promising target for the specific delivery of therapeutic agents.
The therapeutic effect of Apt-αvβ3-siEEF2 is dependent on its specific delivery to tumor cells by Apt-αvβ3, and the cytotoxic effect of siEEF2, which is based on the induction of apoptosis. A control construct combining Apt-αvβ3 with a nonspecific siRNA (Apt-αvβ3-siNON) had no effect on the viability of tumor cells, whereas the requirement for aptamer-based targeting was confirmed by the negligible effect of Apt-αvβ3-siEEF2 on the viability of HEK-293T cells, which express minimal amounts of αvβ3 integrin (Figs. 4 and 5). In this context, we found that the overall toxicity of the construct depended on the quantity of cell surface receptors. Apt-αvβ3-siEEF2 showed twice the toxicity toward U-87 MG cells than SiHa and PC-3 cells, reflecting the higher abundance of αvβ3 integrin on the surface of U-87 MG cells. The relationship between receptor density and cytotoxicity increases the therapeutic potential of this chimeric RNA by building in a failsafe to prevent the targeting of healthy cells with low levels of αvβ3 integrin.
The high concentration of the aptamer–siRNA chimera required for in vitro gene knockdown compared to previous studies (McNamara et al., 2006; Wullner et al., 2008; Dassie et al., 2009) may reflect differences in the target receptors, the internalization pathway of the target receptors, the expression level, and the targeted cell types. The silencing efficiency could be increased by optimizing our aptamer–siRNA chimera, for example, by developing bivalent aptamer-siRNAs (Caswell and Norman, 2006), truncating the aptamer, swapping the position of the s siRNA strands, or adding polyethylene glycol to the 3′-end of the siRNA sequence.
Therapeutic double-stranded RNA can induce an innate type 1 interferon response (Judge et al., 2005; Whitehead et al., 2011), increasing the levels of interferon α and β and thus inducing a panel of antiviral genes (Garcia-Sastre and Biron, 2006). For this reason, we considered several criteria when selecting and modifying the siRNA-aptamer chimeras to avoid an innate immune response, including the removal of Toll-like receptor-7 recognition sequences (5′-UGU-3′ and 5′-GUCCUUCAA-3′) in the RNA chimeras (Heil et al., 2004; SIOUD, 2005; Diebold et al., 2006), and reducing RNA-dependent immunogenicity by incorporating cytosine and uracil residues substituted with 2′-fluorine (SIOUD, 2005). Although our aptamer-siRNA chimeras did not activate a type 1 interferon response in vitro as determined using a human interferon β ELISA kit, in vivo testing is also required to ensure the absence of an innate immune response.
In conclusion, we have demonstrated for the first time that an aptamer binding specifically to αvβ3 integrin can be used to deliver a cytotoxic siRNA to antigen-positive tumor cells. This allows the development of therapeutic modalities composed solely of RNA, reducing production costs and potential immunogenicity compared to protein therapeutics. Aptamer–siRNA chimeras therefore represent a valuable and generally applicable alternative to protein-based drugs that could represent an exciting and efficacious new paradigm for cancer therapy with a significant potential impact in the clinic.
Footnotes
Acknowledgments
The authors would like to thank the German Academic Exchange Service (DAAD) for the financial support of Ahmad Hussain and Dr. Richard M. Twyman for critical reading of the manuscript. Authors' contributions: AFH performed the experiments; AFH., M.K.T., and SB were responsible for project planning; AFH drafted the manuscript; and MKT and SB revised the manuscript.
Author Disclosure Statement
No competing financial interests exist.