
Abstract
Thioacetamido nucleic acids (TANA) contain a backbone modification of dinucleotides consisting of a 5-atom amide linker N3′-COCH2-S-CH2 at thymidine or thymidine–cytidine dimer blocks. Here, the chemical synthesis of the TANA linked 5-methyl-cytidine–cytidine (Mecc) dimer block and its incorporation into the DNA sequence, complementary to human microRNA 34 (miR-34) is described. Further, for the first time, we demonstrate the biological applications of TANA modified oligonucleotides in detection and intracellular knockdown of a cancer related microRNA in comparison with DNA containing locked nucleic acid (LNA) and 2′-O-methyl modifications. The human microRNA miR-34 is a pro-apoptotic microRNA under the transcriptional control of protein 53 (p53). It gets expressed in response to DNA damage and regulates several cell cycle and apoptosis related targets. Here, we show that the TANA modified antisense oligonucleotide binds specifically to miR-34a, allowing its detection using primer extension. We also show that, using the TANA modified antisense oligonucleotide against miR-34a, intracellular levels of miR-34 can be reduced, and consequently, the expression of its target oncogene V-myc myelocytomatosis viral related oncogene, neuroblastoma derived (MYCN) is enhanced. Further, we assessed the toxicity and serum stability of the oligonucleotide to conclude that it is suitable for detection and modulation of the vital biomarker and tumor suppressor microRNA.
Introduction
Several studies have shown that the aberrant expression of microRNAs can lead to simultaneous dysregulation of many target genes, resulting in inappropriate cell death. Usually, microRNAs are repressors; therefore, loss of microRNA expression or activity can result in increased expression of target genes, while induction of microRNAs usually results in decreased target gene expression. Both gain and loss of microRNA expression has been reported in cancer and other diseases. Restoration of microRNA expression to normal levels can be achieved through microRNA mimics or knockdown of the inappropriately expressed microRNA. A major challenge in developing microRNA based therapeutics is the effective and specific knockdown of such microRNAs using antisense technology (Stenvang et al., 2012).
Detection and modulation of microRNAs are therefore essential tools both for fundamental studies and for developing microRNA as potential diagnostics and therapeutics (Van et al., 2012). However, both reliable detection and knockdown of microRNAs are especially challenging due to their small size. Since microRNAs are only 19–24 nucleotides in length, it is difficult to design a highly specific oligonucleotide probe against each microRNA. A widely adopted approach to overcome this challenge is the use of modified backbone oligonucleotides to increase stability of the sense–antisense duplex and thus promote hybridization of probes to the target. Similarly, specific knockdown of microRNAs can benefit from the use of stable backbone modified oligonucleotides. The elevation of melting temperatures of such duplexes also helps in using stringent hybridization conditions during detection of microRNAs in biological systems. Currently, commercially available locked nucleic acid (LNA) antisense oligonucleotides are used extensively for detection and knockdown of microRNAs. However, the development of alternative backbone modifications and the demonstration of their ability to detect microRNAs will help in providing greater choice of probes for microRNA detection and antisense agents for knockdown of small RNAs.
We have earlier reported the synthesis and incorporation of thymidine and thymidine–cytidine dimer blocks connected with a 5-atom amide linker N3′-COCH2-S-CH2 (thioacetamido nucleic acid, TANA) (Gogoi et al., 2007a; Kaur et al., 2009; Gokhale et al., 2010). Using thermal denaturation ultraviolet melting experiments we showed that the introduction of a single thymidine dimer block resulted in selective binding to complementary RNA oligonucleotides and a significant ability to discriminate between complementary DNA and RNA. Although the synthesis of TANA modified DNA analogues containing thymidine dimer, cytidine–thymidine dimer (Gogoi et al., 2007a), and their conjugation with peptides is reported earlier (Gogoi et al., 2007b), this RNA selective DNA analogue has not been used in biological applications such as detection of biologically relevant target RNA molecules or knockdown mediated by antisense molecules. We, therefore, compared TANA to currently used LNA analogue, specifically for detection and knockdown of a specific microRNA implicated in cancer. Here, we report the synthesis of the TANA linked cytidine–cytidine dimer block and show that incorporating this dimer block in modified DNA is an effective alternative to locked nucleic acid modification for both applications, detection of microRNA as well as knockdown of endogenous microRNA in mammalian cells. Further, we show that the knockdown of the microRNA results in the upregulation of the cancer related target V-myc myelocytomatosis viral related oncogene, neuroblastoma derived (MYCN). Mutations in the target region render the MYCN gene recalcitrant to knockdown by the TANA analogue, thus establishing the specificity of the effect. We also show that in the presence of serum, simulating conditions of in vivo delivery, the TANA analogue is as stable as 2′-O-methyl and LNA modifications and is also nontoxic to cells. In summary, we demonstrate the utility of TANA modified probes and antisense molecules as specific agents for detection of a cancer related microRNA biomarker.
Materials and Methods
Detailed methodology for synthesis of TANA modified oligomer is provided in the Supplementary Information (Supplementary Data are available online at www.liebertpub.com/nat).
Cell culture
Human embryonic kidney cell line (HEK293T) and mouse neuroblastoma cells (Neuro 2a) were cultured in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum (Gibco), 1mM sodium pyruvate, 2 mM l-glutamine, and antimycotic antifungal antibiotic (1×). Both the cell lines were cultured at 37°C, 5% CO2 in a humidified incubator according to standard procedures.
Constructs
pMIR-REPORT™ Luciferase (Ambion) was procured commercially. pMIR-MYCN-WT and pMIR-MYCN-MT1&2 constructs were a kind gift from Javed Khan, National Cancer Institute (Wei et al., 2008).
Oligonucleotides and LNA
DNA oligonucleotide and LNA modified oligonucleotides were procured commercially from Sigma and Exiqon, respectively; 2′-O-methyl oligonucleotides and TANA were prepared at National Chemical Laboratory, Pune.
RNA isolation
Total RNA was isolated from cultured HEK293T cells using TRIzol (Invitrogen) according to the manufacturer's protocol. Batches of 50 unfertilized (0 hours post-fertilization) were used for RNA isolation. The RNA pellets were washed with 70% ethanol and air-dried. Pellets were resuspended in nuclease-free water (Ambion). RNA concentration and purity were determined by measuring absorbance at 260 and 280 nm using a spectrophotometer (Eppendorf).
Primer extension
Primer extension was performed from 1 μg of total RNA and primers as mentioned in the figures. Total RNA was annealed with the primer and incubated with a mix of 200 nM dATP, dGTP, dTTP, and α-P-32-dCTP along with M-MuLV reverse transcriptase (New England Biolabs Inc.) for 30 minutes at 42°C for primer extension. Samples were resolved in 20% polyacrylamide gel containing urea (8 M) and the gel was scanned in a Typhoon phosphorimager (Amersham Biosciences).
Real-time PCR analysis of microRNA expression
The microRNA expression was quantified using QuantiMir Real Time PCR assays for miR-34a according to the manufacturer's protocol [System Biosciences (SBI)]. miR-92a was used as endogenous control for expression analysis. Reverse transcriptase (RT) reactions were performed from 2 μg of total RNA with universal reverse RT primers (provided by SBI) at 42°C for 1 hour followed by 10 minutes incubation at 95°C to inactivate the RT enzyme. Real-time PCR was then performed using the reverse-transcribed products and microRNA-specific forward PCR primer for up to 45 cycles. Cycle threshold (Ct) values were analyzed using ΔCt method (Livak & Schmittgen, 2001).
Luciferase assay
For reporter gene assays HEK293T cells were seeded at 18×103 cells per well of a 96-well dish and transfected 24 hours later using Lipofectamine 2000 reagent (Invitrogen) as recommended by the manufacturer. The cells were at 80% confluency at the time of transfection. Each cotransfection reaction contained 200 ng of DNA comprised of pMIR-MYCN-WT construct, backbone modified oligonucleotides at 40nM final concentration, and 1 ng of Renilla luciferase control vector. After 24 hours, cells were lysed and luciferase activity was analyzed using dual-luciferase assay (Promega) in a Top Count luminometer NXT (Perkin Elmer). Firefly luciferase activity was normalized to Renilla luciferase activity and expressed relative to a negative control LNA or TANA.
Cell viability assay
HEK293T and Neuro 2a cells were seeded at 20,000 cells per 96-well dish for cell viability assay. Transfection of HEK293T and Neuro 2a cells was done using Lipofectamine 2000 and Lipofectamine-LTX respectively per manufacturer's protocol. After 24 hours of transfection, 100 μL of cell titre glo buffer was added per well. Luminescence activity (which signifies amount of ATP) was analyzed after 10 minutes of incubation of cells with buffer at 37°C using Top Count luminometer NXT (Perkin Elmer). The amount of ATP is directly proportional to the number of cells present in culture.
Serum stability
Radiolabelled oligonucleotides (0.28 μM) were incubated at 37°C in 10% fetal bovine serum (Invitrogen) diluted in phosphate-buffered saline. Aliquots of 5 μL were withdrawn at different time points and immediately mixed with 5μL of RNA loading dye (Ambion) and frozen. Samples were subjected to electrophoresis in 18% polyacrylamide–Tris borate ethylenediaminetetraacetic acid (TBE) gel under nondenaturing conditions and visualized by Typhoon scanner (Amersham Biosciences).
Results and Discussion
To study the utility of TANA modified oligonucleotides as probes for microRNA detection and antisense agents for microRNA knockdown, we used microRNA-34 (miR-34) as a prototype target microRNA. We chose miR-34 because it has been implicated as a cancer biomarker in multiple studies involving different cancers. Mammalian miR-34 family of microRNAs is comprised of the well-studied, cancer related miR-34a and 2 other closely related sequence homologs, miR-34b and 34c (Hermeking, 2010). The mature miR-34a is highly conserved, with an identical sequence in humans and rodents. It is induced in response to DNA damage in a p53 dependent manner, to promote apoptosis and prevent undesirable cell division. Loss of miR-34a expression has been seen in several cancers. In our own studies (unpublished results) and in reports by Li et al., miR-34a has been shown to be repressed in hepatocellular carcinoma samples (Li et al., 2009). miR-34a is therefore a major effector of p53 mediated apoptosis and regulation of cell division. The expression level of miR-34a is elevated in a mouse model of diabetes and in patients with fatty liver disease (Fu et al., 2012). In a clinical study of circulating levels of microRNAs in patient serum, it was found that along with 6 other microRNAs, miR-34a expression was significantly elevated in newly diagnosed diabetes patients compared with individuals in the prediabetes stage (Kong et al., 2011). Thus, miR-34a is a well-studied microRNA that is a known biomarker in cancer metastasis and liver disease. Further, knockdown of the elevated miR-34a level in diabetes patients is being considered as a potential therapeutic approach.
We first designed short probes that would specifically bind to miR-34a. miR-34a is part of a family of 3 microRNAs—miR-34a, b, and c—wherein miR-34b and c have much lower expression compared with miR-34a. miR-34a also shares partial homology with miR-449a; the seed sequences of miR-34 and miR-449a are identical as shown in Fig. 1 and therefore cannot be used for specific targeting. We therefore used the divergent 3′ end of miR-34 to position the anti-microRNA as shown in Fig. 1A and B. This position is also useful in detecting miR-34 specifically, since it can be extended to the 5′ end using the primer extension method. As shown in Table 1, we designed a 14-nt probe DNA sequence, complementary to the 9th–22nd nucleotide region of miR-34a, (5′ - ACA ACC AGC TAA GA - 3′). Since this sequence warranted it, we synthesized a new TANA linked 5-methyl-cytidine–cytidine (Mecc) dimer block as described in Figs. 1 and 2. The 5′-mercaptoacetic acid derivatives of cytidine were synthesized from corresponding protected nucleoside 2′-deoxycytidine 1 (Fig. 2). The 5′-tosyl derivative, 1a, was obtained by selective tosylation of 1. The selective tosylation was achieved under high dilution conditions and by controlled addition of tosyl chloride at 0°C. Mercapto nucleophile generated in-situ, by reaction of NaH and ethyl mercaptoacetate, was used to substitute the tosyl group to get mercapto derivative 1b. The 3′-OH group was further protected as tert-butyldimethylsilyl (TBDMS) ether by reaction with TBDMS-Cl in dimethylformamide to get 1c. While attempting ester hydrolysis for compound 1c using LiOH (Gogoi et al., 2007b), benzoyl protection of cytosine exocyclic amino group was also hydrolyzed. Hydrolysis of the ester group in 1c was attempted with different conditions like triethyl amine/water, 1,8-diazabicycloundec-7-ene/water, but these conditions were not successful for hydrolysis of the ester group. The selective hydrolysis of the ester group in 1c was effectively achieved with 1N NaOH in tetrahydrofuran/water mixture at 0°C. Under these conditions, hydrolysis of the ester group was achieved keeping benzoyl protection of the cytosine base intact to give product 1d. TANA linked Mecc dimer block 5, suitable for incorporation in solid phase oligonucleotide synthesis, was synthesized as shown in Fig. 3. The 5′-mercaptoacetic acid-2′-deoxycytidine derivative 1d was coupled with 2′-3′-dideoxy-3′-amino-5-methyl cytidine 2 (Gokhale et al., 2010) with moderate yield. The protected dimer 3 was purified by column chromatography. Compound 3 on desilylation gave compound 4, which on subsequent phosphitylation gave the phosphoramidites 5. The phospharamidite derivative 5 (Mecc) was used to incorporate Mecc dimer block into the DNA sequence using automated DNA synthesizer.

Sequences of microRNA (miR)-34 and miR-449 family and their binding patterns with antisense oligonucleotides: miR-34 and miR-449 family members share an identical seed sequence. The seed sequence of the microRNAs is represented in bold underlined letters. The predicted binding pattern of the locked nucleic acid (LNA)/2′-O-methyl
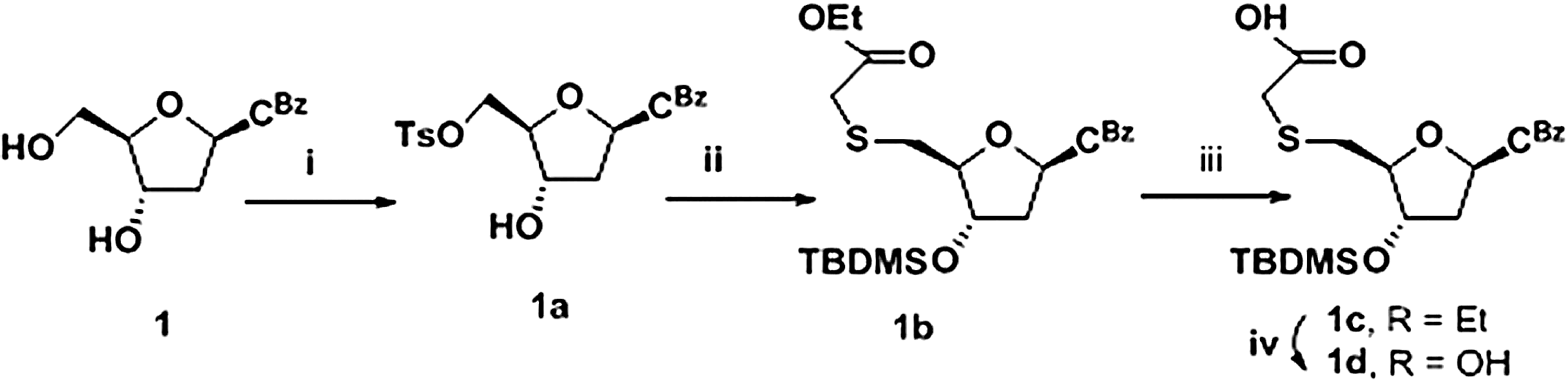
Reagents and conditions:
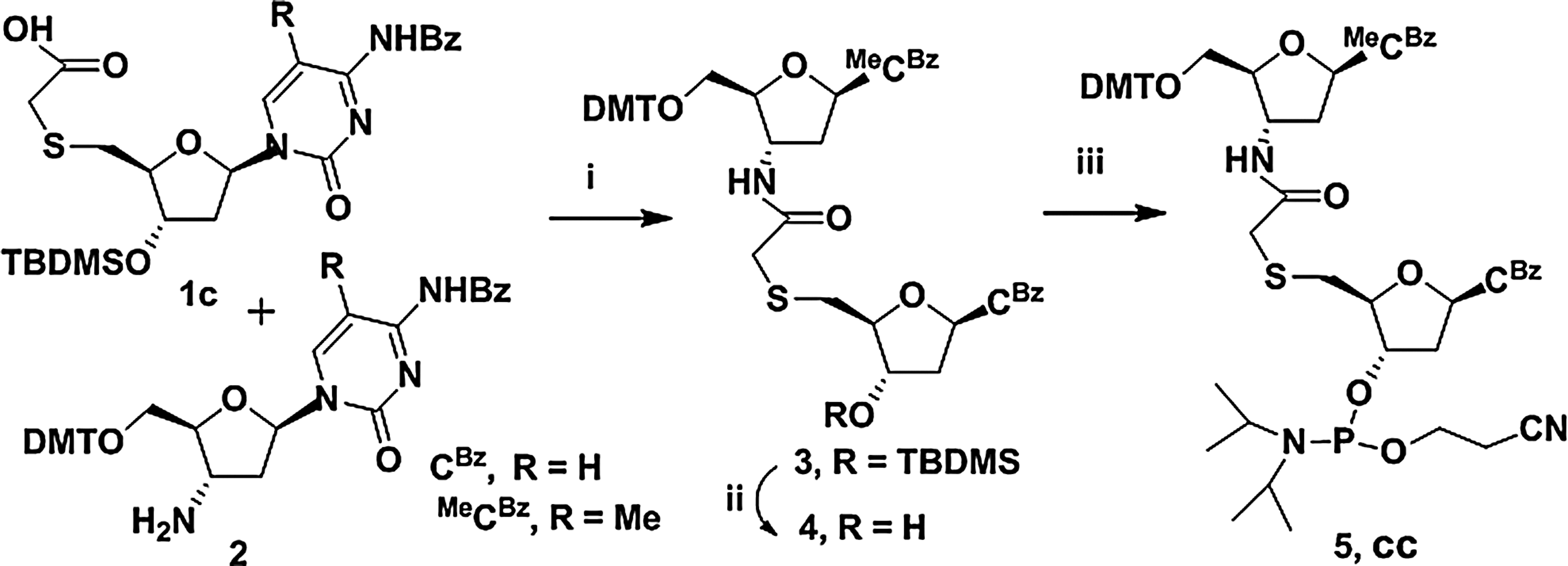
Reagents and conditions
DNA oligo is unmodified. The modification in TANA oligo is present in between the small lettered nucleotides; underlined nucleotides represent the modified bases in LNA; and nucleotides that are preceded by letter “m” represent the modified bases in 2′-O-methyl.
Since we have earlier seen that multiple TANA modifications reduce the affinity for the target (Kaur et al., 2009), we introduced a single cytidine dimer block in the only available cytidine–cytidine dinucleotide site in the probe (TANA- 5′ACA AMecc AGC TAA GA) encompassing the fifth and sixth positions. We also created four probes each with LNA modifications and 2′-O-methyl modifications. The four LNA modified probes, for instance, had an increasing number of modified nucleotides and were referred to as LNA-1,2,3 and 4 (Table 1). Initially, we used all of the oligonucleotides to study the relative efficiency in detection of miR-34a with multiple LNA or 2′-O-methyl modifications in comparison with unmodified DNA. MicroRNAs are routinely detected in laboratory studies using northern blotting and hybridization; however, real-time polymerase chain reaction (PCR) or other sensitive methods are required for the detection of microRNAs in patient samples due to the limited quantity of the samples available. We used primer extension as an assay for microRNA because it is quantitative, allows direct visualization of the product, and does not require any specialized equipment or reagents. In primer extension, the endogenous target microRNA, present in the total RNA pool, is allowed to anneal with a complementary probe. The method relies on “extension” of the probe in the duplex by reverse transcriptase. Visualization of the product is achieved by incorporating radioactively labeled nucleotides during the extension step or by using prelabeled probes. The reaction products are subjected to polyacrylamide gel electrophoresis or capillary gel electrophoresis. We have used this method extensively (Ahluwalia et al., 2008) and found that the sensitivity is sufficient to detect microRNAs having low expression levels, from 1 to 5 μg total RNA, without any requirement for small RNA enrichment or size fractionation steps. Here we used RNA from zebrafish embryos for initially studying the ability of the probe to detect the target microRNA. We found that, in agreement with the literature, increasing the number of LNA and 2′-O-methyl modifications in the backbone resulted in better detection sensitivity of miR-34a (Fig. 4). The single TANA modification was sufficient to confer sensitivity comparable to a single LNA or 2′-O-methyl modification. The product was specific and of the expected size (22 nt), since we did not see any additional bands. The unmodified 14-nt probe with the native DNA backbone was not capable of detecting miR-34a under these conditions. Having established that the single TANA modification was sufficient for miR-34a detection and was comparable to single modifications of LNA and 2′-O-methyl in the probe, we carried out further experiments with these probes. We also used unrelated probes with the same number of modifications as a negative control, to rule out non-specific effects on gene expression.
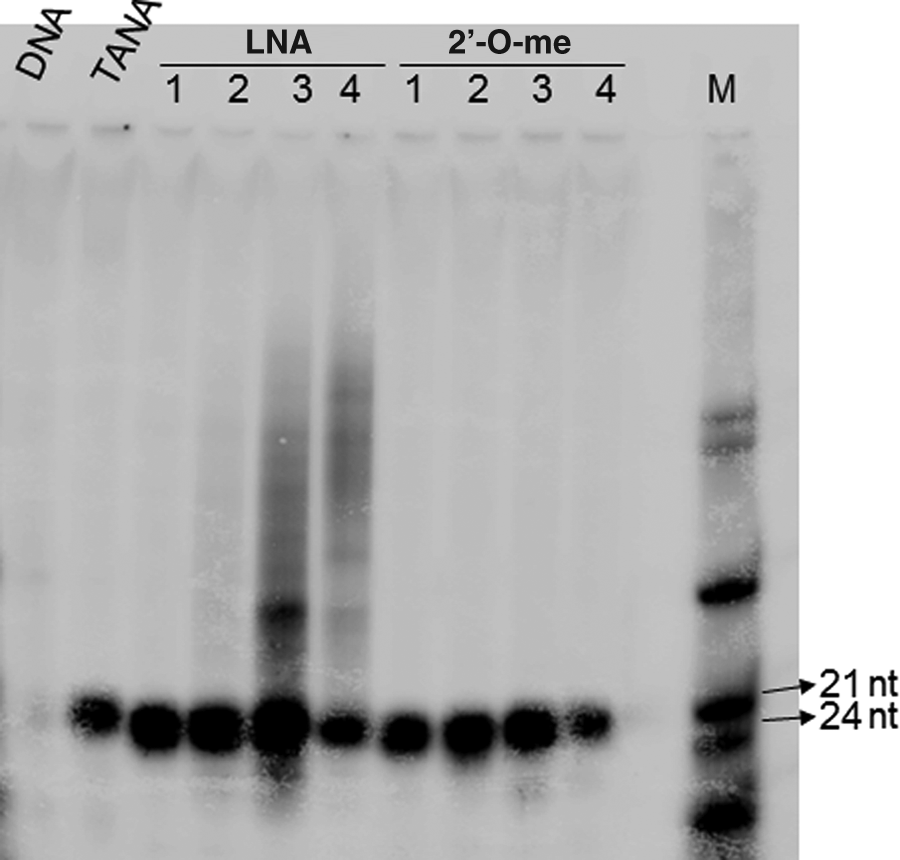
Effect of backbone modification on detection of miR-34a by primer extension with oligonucleotide primers, with modifications as indicated in Table 1, were used to detect miR-34 in total RNA from zebrafish embryos. Total RNA was denatured by heating and re-annealed to radioactively labeled primers. Taq DNA polymerase was used to extend the primer to the full length of the template. The reaction was then stopped and the contents electrophoresed on an 18% polyacrylamide gel. Bands were visualized using phosphorimager. All of the oligos except the DNA oligo were able to detect miR-34a during the primer extension. One TANA modification was found comparable to a primer with a single LNA or 2′-O-methyl (2′me) modification. A radioactively labeled mix of primers (M) was used to confirm the expected size of the product.
Next, we tested the ability of these oligonucleotides to act as antisense knock down agents in the cell. We used cultured HEK293T cells since miR-34a is expressed highly in this cell line. We transfected cells with the negative control oligonucleotide, oligonucleotide with 1 backbone modification with LNA, TANA, or 2′-O-methyl oligonucleotide using lipofectamine. We transfected the oligonucleotides at 40 nM concentrations because we have previously shown that at this concentration, the LNA modified oligonucleotides had the most effective knockdown efficiency in cell culture based experiments (Ghosh et al., 2008). The expression of mature miR-34a was monitored using a commercially available real-time PCR based microRNA assay (see methods section). We found that the expression of miR-34 was reduced by 66% by all the antisense oligonucleotides (Fig. 5). TANA was comparable to LNA and 2′-O-methyl oligonucleotide with similar number of modifications in the level of knockdown achieved. However, a caveat in such analyses is that the residual antisense oligonucleotide may interfere with the microRNA detection, making it impossible to attribute the knockdown to a purely intracellular effect.
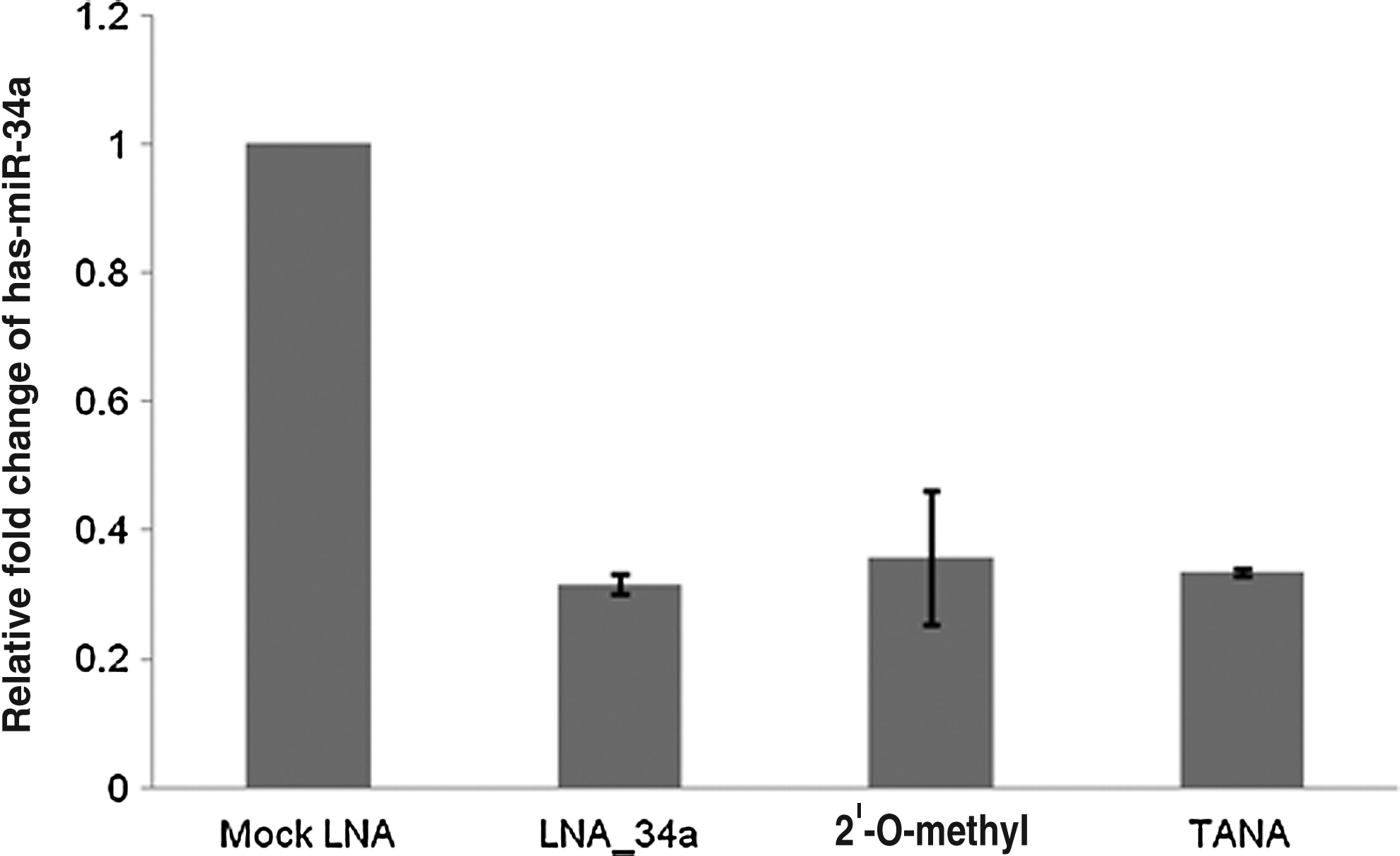
Effect of backbone modification on knockdown of miR-34a. Cells were transfected with oligonucleotides as indicated. After 24 hours of transfection, total RNA was collected and the change in the endogenous expression level of miR-34a was monitored by quantitative real-time PCR. TANA modification was as effective as LNA and 2′-O-methyl modifications in down regulating miR-34a.
Next, we assessed the cellular toxicity of the antisense oligonucleotides used in two different cell types, Neuro 2a and HEK293T cells. We used a commercially available luminescence based assay for measuring ATP levels to detect viable cells, based on the rationale that ATP levels are directly proportional to number of viable cells. This assay is more sensitive than other conventional assays like MTT assay for cell viability. We saw that the assay reliably detected rapid cell death following a brief exposure to 50% ethanol (data not shown). Irrespective of the modifications, none of the antisense oligonucleotides affected viability of either of the cell lines; hence, we ensured that TANA modification has no significant cellular toxicity at the concentration indicated above sufficient to achieve a 66% knockdown (Fig. 6).
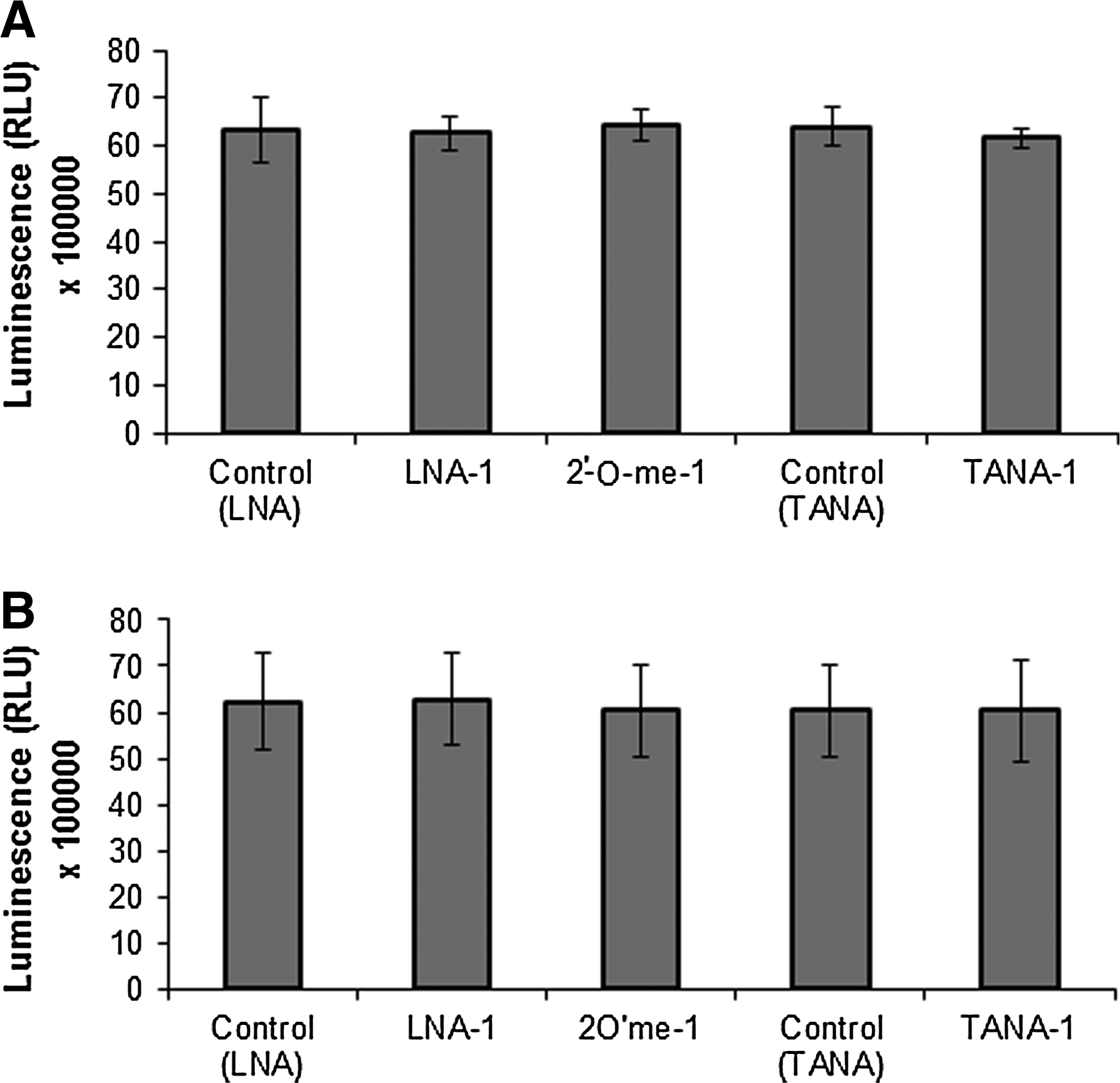
Toxicity of backbone modified oligonucleotides. Viability of cells after being transfected with backbone modified oligonucleotides was studied by measuring ATP levels represented as relative luminescence units in
MicroRNAs in the cell are associated with a multi-protein complex known as the microRNA induced silencing complex, which contains effector argonaute proteins. The microRNA directs the complex to target messenger RNAs with regions partially complementary to the microRNA, wherein the effector proteins either cleave or block translation of the target mRNA depending on the degree of complementarity between the two. The final consequence of both target degradation and translational block is the down-regulation of the target protein. Conversely, knockdown of microRNA expression is expected to result in elevated target gene expression. The expression of MYCN, an oncogene known to play a critical role in the tumorigenesis of neuroblastomas, is known to be regulated by miR-34a (Wei et al., 2008). We therefore used the miR-34 target region in the 3′ untranslated region (UTR) of the MYCN transcript to ensure that the knockdown of the microRNA results in the expected upregulation of the microRNA target gene expression. We used a fusion of the luciferase reporter gene with the 3′UTR of the MYCN gene having 2 binding sites for miR-34a, to monitor the effect of microRNA knockdown on target gene expression. We found that TANA modified antisense oligomers showed 27% elevation in the target expression as measured by relative luciferase expression levels (Fig. 7). This is in agreement with other reports in the literature that individual microRNAs usually bring about only 30%–60% down regulation of targets, acting to fine-tune gene regulation.
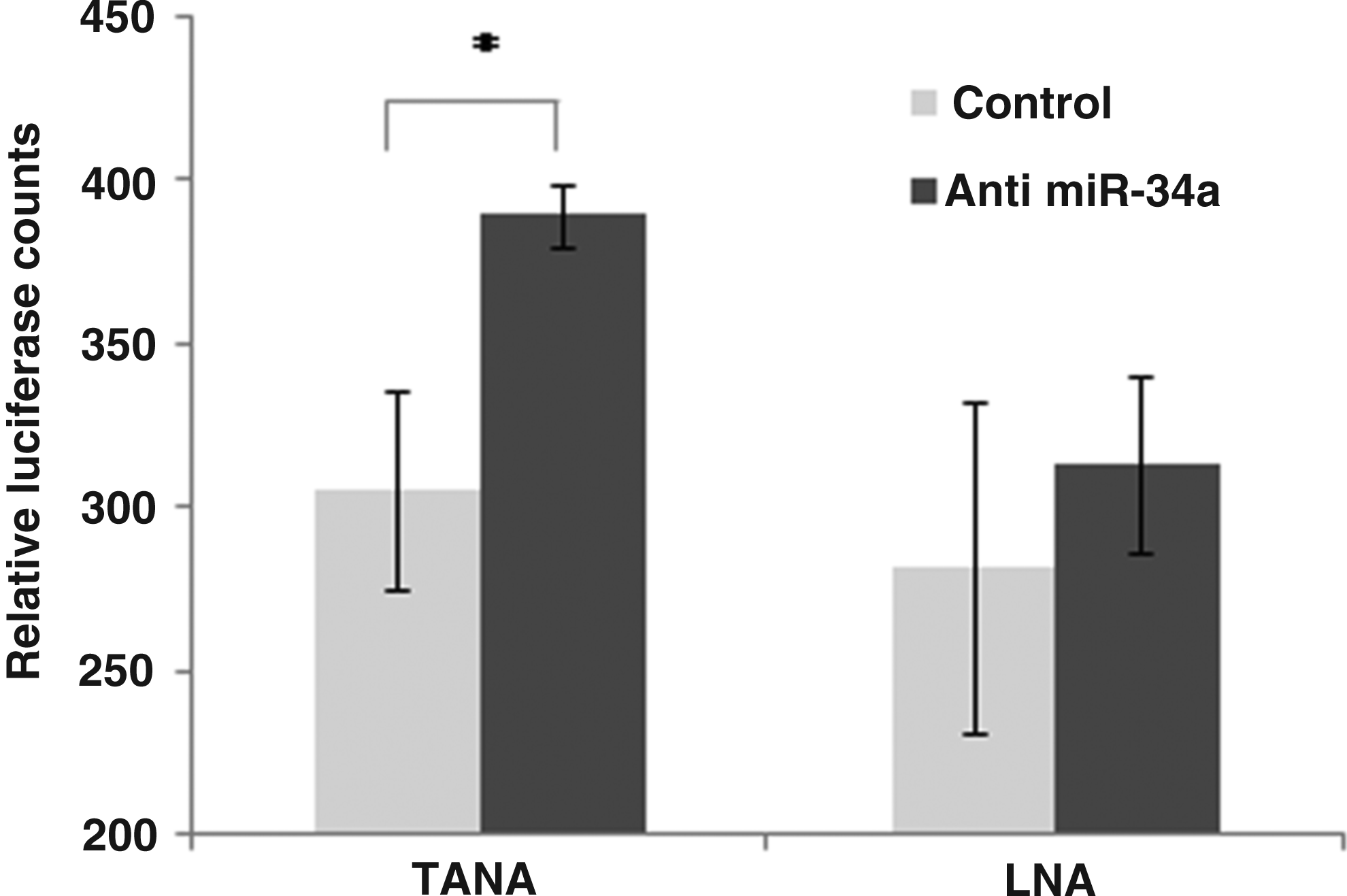
Recovery of target gene expression following microRNA knockdown. Luciferase activity from a Luciferase gene fused to the 3′ untranslated region (UTR) of MYCN gene was used as an indicator of miR-34 levels in the cell (black bars). The plasmid containing the miR-34a target was cotransfected with oligonucleotides carrying a single LNA modification (LNA-1) or TANA. Unrelated LNA or TANA modified oligonucleotides were used as control. In all the experiments, transfection and luciferase assays were performed as technical replicates; results reported here are from 2 biological replicates for LNA and 3 biological replicates for TANA (*represents P-value≤0.01). MYCN, V-myc myelo-cytomatosis viral related oncogene, neuroblastoma derived.
Lastly, we studied the serum stability of TANA modified oligonucleotide comparing it to LNA and 2′-O-methyl modified oligonucleotide in order to test the stability of these backbone modified oligonucleotides in vivo. We incubated prelabeled oligonucleotides with fetal calf serum and monitored its stability over time using polyacrylamide gel electrophoresis. We found that the oligonucleotides having similar modifications of all three types (LNA, TANA, and 2′-O-methyl) had comparable serum stability (Fig. 8), thus suggesting that TANA is as effective, stable and sensitive antisense agent as the currently commercially available LNA and 2′-O-methyl analogues.
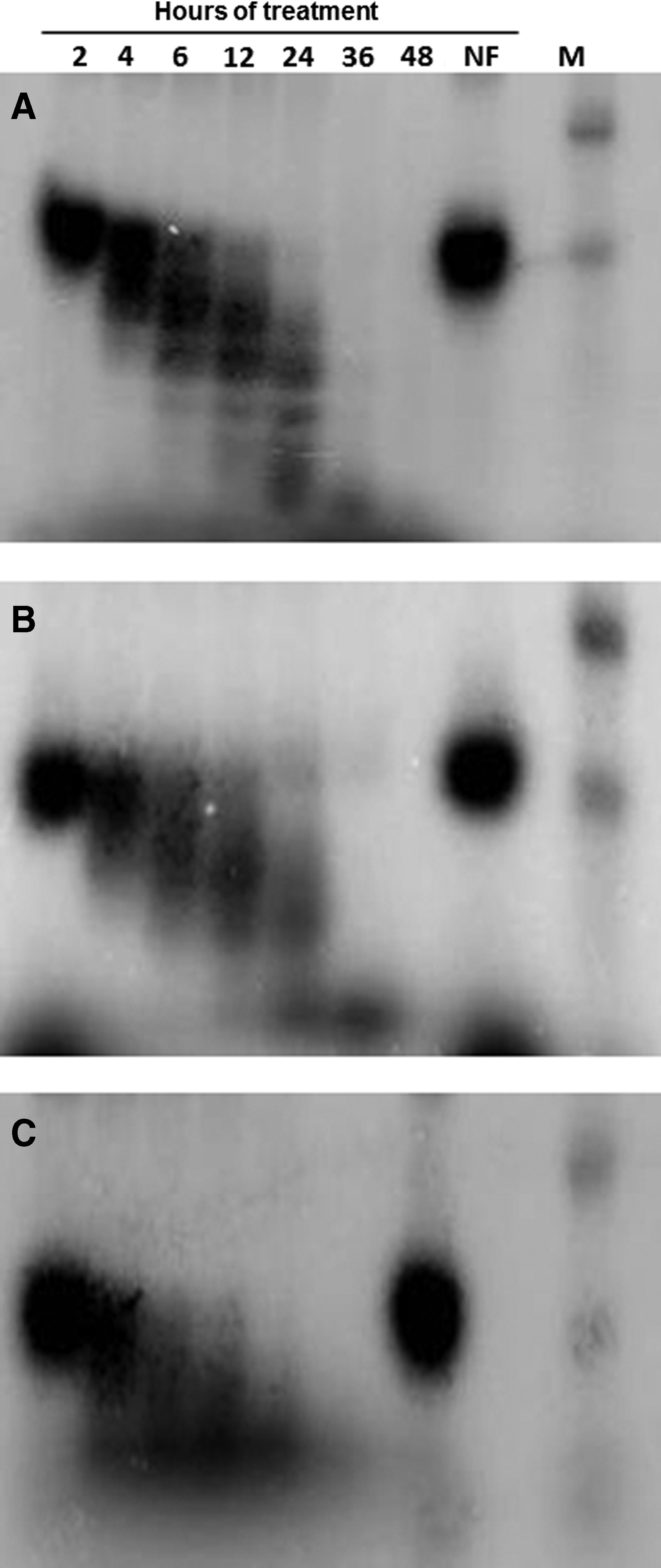
Serum stability of oligonucleotides with a single modification of TANA
Thus, the synthesis of a novel Mecc dimer block for incorporation into oligonucleotides and its successful use in antisense mediated microRNA knockdown is described. We assessed the ability of TANA modified oligonucleotides to hybridize to microRNA, knock down microRNA, and restore the expression of the target of the microRNA, as well as its stability in serum and toxicity levels in human cell culture models. In all the tests, the performance of TANA modified oligonucleotides was only comparable to LNA modification. LNA modified oligonucleotides being relatively expensive, the alternative modified and effective anitsense oligonucleotide is likely to make positive impact.
It is interesting to note that both LNA and TANA modified antisense molecules not only bind to the microRNA and interfere in its binding to target, but also lead to the degradation of the microRNA, resulting in a reduced level of expression of the microRNA itself over longer periods of time. In the future, it will be interesting to identify cellular factors responsible for such degradation. The conjugation of TANA modified oligonucleotides to peptides has already been demonstrated (Gogoi et al., 2007b). Cell penetrating peptides have been used to deliver small interfering RNA to specific tissues. In the future, TANA probes conjugated to cell penetrating peptides can be used for specifically knocking down microRNA in some cells. In summary, we present a first report of a systematic comparison of a TANA modification with LNA to establish it as a useful probe for specific detection and knockdown of microRNAs.
Footnotes
Acknowledgments
We acknowledge funding support from the Council of Scientific and Industrial Research, India through the project “Comparative Genomics of non-coding RNA” (NWP0036). KS was supported by a senior research fellowship from the Council of Scientific and Industrial Research, India. The authors would also like to thank Souvik Maiti for scientific suggestions. The MYCN 3′UTR carrying plasmid construct used in the study was a kind gift from Javed Khan, National Cancer Institute, Gaithersburg, Maryland.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.