SharpPhillip A.
Massachusetts Institute of Technology
RNA Biology and Therapeutic Innovations
The past decade has produced an explosion in recognition of the roles of RNA in biology. The discovery of both RNA interference (RNAi) and microRNAs revealed a previously unappreciated world of small RNAs in the regulation of genes. Most of this regulation is post-transcriptional and targetable by agents introduced into the cytoplasm of cells. Small RNAs also can function in the nucleus to regulate transcription by controlling chromatin structure. Deep RNA sequencing has further detected many long non-coding RNAs that may also regulate cellular processes and gene expression. In many examples, these are transcribed in a divergent fashion from the promoters of protein-coding genes or from enhancers controlling the expression of these genes. Most of these thousands of non-coding RNAs are expressed at the level of a few copies per cell and their functions are unknown. Nevertheless, a few non-coding RNAs, such as XIST, are present at higher copies per cell and have known roles in cell biology. Clearly the functional roles of this large set of long non-coding RNAs need to be better understood before considering them for therapeutic interventions. The complexity of RNA biology in the form of alternative RNA splicing was appreciated long before the discovery of small non-coding RNAs. Alternative RNA splicing is essential for normal physiology and is frequently altered in disease processes. Over 90% of all genes in human cells are alternatively spliced in different cell types. Mutations in splicing sequences can shift alternative splicing patterns and can be targeted by therapeutic agents to suppress disease phenotypes. RNA processing signals are coupled to both transcriptional processes in the nucleus and translational processes in the cytoplasm. Shifts in the recognition of these RNA processing signals control mRNA structure and thus regulation by interacting miRNAs and proteins. These same signals probably also control the expression of long non-coding RNAs and, in fact, may account for the differences in their abundance and functions. Although the important roles of RNA in biology have been appreciated for sometime, the technology to manipulate RNAs in vivo with synthetic oligonucleotides is relatively new. This technology represents a new age in pharmacology with characteristics that make it attractive for personalized medicine. Great progress has been made over the past few years but this is only the beginning.
MaraganoreJohn
Alnylam Pharmaceuticals
RNAi Therapeutics: State of the State
Therapeutic agents that act through the RNA interference (RNAi) pathway are specific and potent inhibitors that may be designed to disease pathways previously considered “non-druggable”. Numerous proof-of-concept studies in animal models of human disease demonstrate the broad potential of RNAi. Recently, Alnylam has demonstrated both proof-of-concept and proof-of-mechanism of RNAi therapeutic agents in clinical trials in disease areas such as liver cancer, respiratory syncytial virus, hypercholesterolemia, and transthyretin-mediated amyloidosis. The major challenge for the successful development of systemically delivered RNAi therapeutics has been to identify delivery approaches that could be translated into the clinic. Tremendous progress has been made in this area, and importantly, clinical trials are underway with several RNAi therapeutic candidates using lipid nanoparticles (LNPs) for intravenous administration. At Alnylam, we have also discovered an siRNA-GaNAc conjugate platform that enables subcutaneous delivery of RNAi therapeutics targeted against genes expressed in hepatocytes. Using these complementary approaches, Alnylam is advancing several RNAi agents specific for liver targets to address genetically defined diseases with high unmet medical need. The recent positive clinical results with ALN-TTR, an RNAi therapeutic for the treatment of transthyretin-mediated amyloidosis, a fatal, autosomal dominant, multisystem disease caused by abnormal extracellular deposits of transthyretin amyloid fibrils, and pre-clinical data of ALN-AT3 to target antithrombin for the treatment of hemophilia will also be discussed. Success at Alnylam in imparting drug-like properties to siRNAs by chemical modifications and in delivering these agents systemically emphasizes the tremendous progress made in this relatively young field.
CrookeStanley T.
Isis Pharmaceuticals
Antisense Technology: Past, Present, Future
Progress in antisense technology has identified multiple post hybridization mechanisms that have been successfully exploited to produce pharmacological effects in cells, animals and humans. Advances in understanding the pharmacokinetics and toxicological properties of various types of oligonucleotides have also been reported. Together these data support optimism that RNA based therapeutics may bring value.
Rare Genomics Institute
Tumor-targeted nanoparticles that exploit oncomiR addiction for cancer therapy
The purpose of this study was to develop a targeted therapeutic nanoparticle system that delivers anti-miRs to inhibit the function of oncogenic microRNAs. Ubiquitously expressed in different cells and tissues, microRNAs (miRNAs) have critical roles in various diseases; dysfunction of miRNAs has been linked to the onset and progression of many human cancers. In particular, miR-155 and miR-21 exhibit differential expression levels in certain cancers resulting in a demonstrated capability to affect cellular transformation and carcinogenesis by acting as miRNA oncogenes, or oncomiRs. In fact, cancers can become addicted to miR-155 and miR-21 such that oncogenesis is dependent on the overexpression of the oncomiR, and withdrawal of the oncomiR leads to cancer cell death. The addictive nature of miR-155 and miR-21 makes them potent targets for anti-cancer therapy.
Current strategies for inhibiting miRNAs are limited in vivo by safety and target specificity concerns. Due to their stability and ability to bind complementary nucleic acids with high affinity, peptide nucleic acids (PNA) are effective anti-miRs, i.e. oligos that bind to and inhibit miRNAs. However, PNAs do not efficiently target specific tissues and enter cells without a delivery vector. We have shown that PNAs can be loaded into polymer nanoparticles (NPs); furthermore, this charge-neutral oligo cargo precludes the addition of cationic counter-ions which contribute to the toxicity and unfavorable biodistribution profiles that limit many cationic gene therapy delivery systems. The efficacy of these NPs can be significantly enhanced by coating their surface with functional molecules to help them overcome delivery barriers. Previously, we functionalized anti-miR-loaded NPs with PEG and a cell-penetrating peptide to improve nanoparticle circulation and tumor cell uptake in hard-to-transfect lymphocytes. In this study we coated NPs with PEG and a novel molecule which preferentially targets to acidic tissues when administered systemically, while avoiding the liver, which is a common sink for systemically administered therapies. Encapsulating PNA anti-miRs against miR-155 and miR-21 into these NPs represents a new delivery approach toward inhibiting aberrantly expressed miRNAs in tumors.
Here we will discuss the tissue biodistribution of these targeted NPs, and the therapeutic response to anti-miR-155 and anti-miR-21 delivery in cell culture and mouse models of breast cancer and inducible models of oncomiR addicted lymphoma. Additionally, we will outline the impact of anti-miR-155 and anti-miR-21 therapy on biological pathways involved in oncomiR addiction such as apoptosis, cellular proliferation, and angiogenesis. Lastly, we will detail the synergism of combination therapies using NPs to deliver both anti-miRs and chemotherapeutics, since both miR-155 and miR-21 have been shown to be involved in the development of chemoresistance. OncomiRs have recently established a new paradigm for anti-cancer gene therapy. This work introduces a tumor-targeted nanoparticle system that exploits these lynchpin molecules as therapeutic tools. In addition to cancer, the versatility of this delivery platform may guide the engineering of anti-miR-based therapeutics for a myriad of miRNA-regulated diseases.
Alnylam Pharmaceuticals
Long Noncoding RNAs Link Transcriptional Regulation of Inflammatory Pathway Genes
Although many long noncoding RNAs (lncRNAs) are discovered, their mechanism and function remain obscure. We identify lncRNAs as critical regulators of basal and inducible COX-2 transcription. lncRNAs overlapping the COX-2 promoter are targets for ribonucleoprotein complexes containing complementary small RNAs and RNAi factors, argonaute-2 and GW182. Recognition of lncRNAs by the RNAi machinery triggers recruitment of RNAP2 and transcription factors, enhances COX-2 expression, counteracts repression by dexamethasone, and leads to >100-fold super-activation of COX-2 expression when combined with pro-inflammatory stimuli.
Activation alters histone marks and requires the multifunctional scaffolding protein, WDR5. Gene looping allows long distance interactions between the promoters of COX-2 and PLA2G4A, an adjacent inflammatory pathway gene. Endogenous miR-589 targets two adjacent sequences within a promoter lncRNA to regulate basal COX-2 and PLA2G4A expression. Our data demonstrate that a noncoding RNA network at the COX-2 promoter is controlled by miRNAs and organizes a novel multi-gene inflammatory response pathway.
MeisterGunterBeitzingerMichaela
University of Regensburg
Highly efficient siRNAs with very low off-target effects
Short interfering RNAs (siRNAs) are a potent and widely used tool for sequence-specific gene knock down known as RNAi. siRNAs are incorporated into RNA-induced silencing complexes (RISCs) and guide them to complementary target sites located on other single stranded RNA molecules. RISC subsequently cleaves the target RNA. In a typical RNAi experiment, double stranded siRNAs are transfected and the guide strand binds to a member of the Argonaute (Ago) protein family widely viewed as the core of RISC. In mammals, Ago2 is the only Ago protein that possesses endonuclease activity and is therefore the most important Ago protein for RNAi. However, siRNAs hybridize to partially complementary sequences as well and cause off-target effects, which frequently result in severe problems in RNAi experiments. Here we report the distribution of different siRNA sequences between the human Ago proteins, when large pools of siRNAs are transfected simultaneously. Furthermore, we have investigated siRNA activities when transfected as individual siRNAs or as part of siRNA mixtures. Based on our results, we have developed a novel method that solves the RNAi off-target problem and allows for a rapid generation of highly potent siRNAs with very low off-target effects.
DasSubha R.ParedesEduardoGrahacharyaDebasish
Carnegie Mellon University
Lariat and Mini-lariat RNA: Synthesis and Debranching-Dependent RNA Interference
During the process of splicing that joins exons to generate the correct messenger RNA, introns are removed as lariat structures. These lariat RNAs have an invariant adenosine branch-point residue which is linked at the 2′-position to the 5′-end of the RNA sequence that corresponds to the conserved residues of the intron. Subsequently, the lariat RNA is the substrate for lariat debranching enzyme (Dbr1) that specifically cleaves the unique 2′-5′-phosphodiester bond in the lariat RNAs before they are used in other cellular processes. Recently it was reported that debranching of the 2′,5′-phosphodiester linkage in some lariats (of mirtrons) can generate microRNAs that bypass Drosha. Here we describe the synthesis of lariat RNAs in both solution and solid phase. We have developed chemistry for backbone branching at the 2′-position in RNA and we enhance this with the use azide-alkyne cycloaddition (click-chemistry) – both in solid phase and in solution on naïve RNA (with free 2′-hydroxyls) to furnish branched RNAs in which the loop is closed to complete the lariat structure. This chemical ligation yields a triazole linked lariat RNA with the location of the triazole linkage either at the branch-point 2′-position or downstream. Additionally we also synthesize directly in the solid-phase, mini-lariat RNAs that correspond to the silencing competent guide strand of siRNAs. These mini-lariats have all native phosphate linkages or include a backbone triazole and the structures do not have a 5′-terminus, thereby requiring only a 3′-modifcation for exonuclease resistance. These synthetic lariats or mini-lariats are substrates for debranching in vitro. Debranching serves to linearize the RNA and concomitantly generates a 5′-phosphate terminus. We therefore tested the mini-lariats directly for in vivo RNA silencing and compared their effectiveness compared to single stranded RNA as well as duplex siRNA. Our positive results suggest that synthetic mini-lariat RNAs may represent a significant new class of drosha and dicer independent RNA silencing agent.
HirschMarkusHelmMark
Institute of Pharmacy and Biochemistry - University of Mainz
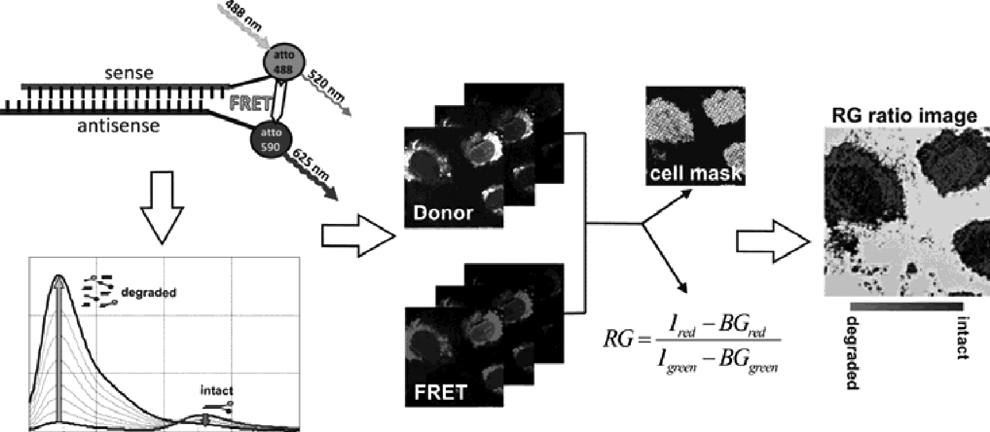
siRNA dynamics – What happens inside a cell? About uptake, release, degradation and stability
RNAi is a powerful tool for analyzing gene function and promises high potential for therapeutic application. While in cell culture models efficient knockdown rates can be achieved, applications in vivo remains challenging. Especially the delivery of siRNA into the target cell and a progressing degradation of siRNA, applied to the target organism or cell, are the major hurdle for therapeutic application.
Labeled siRNA can be used for both observing cellular localization and analyzing the integrity level of the applied siRNA. Two dyes on the siRNA duplex, atto488 at the 3′-end of the sense strand and atto590 at the 5′-end of the antisense strand, create a robust system for determining the integrity level via FRET [1]. The ratio of FRET to Donor emission serves as a sensitive classifier to determine the integrity of the duplex [2,3]. With this system siRNA degradation of less than 5% can be determined in either the cuvette or in living cells.
Results show that siRNA duplexes seem to stay intact for several days, serving as a deposit for prolonged RNAi. The nature of this deposition is still unclear but the area around the nucleus or lysosomes are the best candidates. Upon the release of siRNA from lipoplexes, uptake into the nucleus is observed. The whole release process occurs spontaneous and changes in cellular distribution can be monitored within few minutes. Additionally, the released siRNA is cleared from the cytoplasm over time, leaving only small aggregates of siRNA inside the cell.
HirschM, StrandD, HelmM. 2012. Dye selection for live cell imaging of intact siRNA. Biol Chem., 393:23–35.a-14
JärveA, MüllerA, KimI-H, RohrK, MacLeanC, FrickerG, MassingU, EberleF, DalpkeA, FischerR, TrendelenburgM, HelmM. 2007. Surveillance of siRNA integrity by FRET imaging. Nucleic Acids Research, 35:e124.a-15HirschM, KimI-H, JärveA, FischerR, TrendelenburgM, RohrK, MassingU, HelmM. 2011. Tracing of siRNA inside cells by FRET imaging. Neuromethods:RNA Interference Techniques. HarperS. 141–57. Humana Press.a-16JulianoRudy1MingXin1FisherMichael1NoelRomain2CintratJean-Cristophe2BarbierJulien2CarverKyle1BaumanJohn1NakagawaOsamu1GilletDaniel2
UNC
CEA
Improving the Pharmacological Effects of Oligonucleotides by Enhancing Endosomal Escape
Achieving strong pharmacological effects with oligonucleotides is impeded by poor access of these molecules to their sites of action in the cytosol or nucleus. Lipid or polymeric delivery systems have been only partially successful in addressing this issue. Here we describe two novel alternative approaches for enhancing oligonucleotide effectiveness through promotion of escape from endosomal compartments. The first approach involves the design and synthesis of peptide-oligonucleotide conjugates that contain both a receptor-specific targeting moiety and an endosomal escape moiety. Using conjugates of splice switching oligonucleotides (SSOs) we show that inclusion of the escape moiety provides a substantial enhancement of effect as compared to conjugates containing only the targeting moiety. The second approach involves use of non-toxic small molecules that promote the escape of oligonucleotides from non-lysosomal endomembrane compartments. We have identified molecules that can substantially enhance the effects of both SSOs and single-stranded antisense oligonucleotides in cell culture. We have also shown that one of these compounds can enhance SSO effects in vivo.
MacRaeIan
The Scripps Research Institute
Structural and Mechanism of Human Argonaute-2
Argonaute proteins form the functional core of the RNA-induced silencing complex (RISC) and related gene-silencing complexes that mediate RNA silencing in eukaryotes. We determined the 2.3 Å resolution crystal structure of full-length human Argonaute2 (Ago2) bound to a heterogeneous mixture of single-stranded guide RNAs. Ago2 has a bi-lobed structure, similar to that observed in its bacterial cousins, decorated with extended loops and appendages. The 5′ end in the guide RNA is anchored in the Mid domain, with the solvent-exposed Watson-Crick edges of nucleotides 2 to 6 positioned in an A-form conformation for base-pairing with mRNA targets. The A-form structure is disrupted by a kink between nucleotides 6 and 7. Tandem tryptophan binding pockets in the PIWI domain define a likely interaction surface for GW182 and other glycine-tryptophan rich proteins.
ButoraGabor
Merck
Chemically Modified Nucleosides for RNAi: A Platform Approach
RNA interference (RNAi) is an evolutionarily conserved cellular mechanism for gene suppression found in fungi, plants and animals. Short interfering RNAs (siRNA) trigger a specific cleavage of complementary messenger RNAs (mRNA) via endogenous cellular machinery. siRNAs are naturally produced by an enzyme-mediated cleavage of large double stranded RNAs, but can also be introduced into cells by pharmacologically relevant exogenous pathways. In order to realize the immense therapeutic potential of RNAi, many properties of siRNAs, including potency, stability and immunogenicity need to be optimized. Chemical modifications of naturally occurring nucleosides offer an excellent opportunity to address these challenges, however the synthetic complexity associated with the preparation of these building blocks has so far hampered such efforts. In this presentation we wish to describe a simplified platform approach for siRNA optimization. This new methodology relies upon rapid and efficient chemical modification of nucleosides containing heterocycles capable of Watson-Crick base pairing with naturally occurring canonical bases. Application of this high-throughput, lower-cost methodology allows for the hypothesis driven, rational design of potent, immunologically silent and stable siRNAs.
MatsudaAkiraTakahashiMaymuiMinakawaNoriaki
Hokkaido University
2′-O-Me-4′-thioRNA as a Potential Use for Oligonucleotide Therapeutics
2′-OMe-4′-thioRNAs have high hybridization affinity for complementary RNAs and significant resistance toward degradation not only by nucleases, but also in human plasma1). These results prompted us to develop chemically modified siRNAs using 2′-OMe-4′-thioribonucleosides for therapeutic application. Effective modification patterns were screened using a luciferase reporter assay. The best modification pattern of siRNA, which conferred duration of the gene silencing effect without loss of RNAi activity, was identified. Quantification of the remaining siRNA in HeLa-luc cells using a Heat-in-Triton (HIT) qRT–PCR revealed that the intracellular stability of the siRNA modified with 2′-OMe-4′-thio- ribonucleosides contributed significantly to the duration of its RNAi activity2).
TakahashiM.et al.Nucleic Acids Res., 40:5787. 2012.a-28TuschlThomasWilliamsZevBen-DovIddo Z.RenwickNeilCekanPavolHafnerMarkus
Laboratory of RNA Molecular Biology, Howard Hughes Medical Institute, The Rockefeller University
MicroRNA Quantification in Tissue Sections and Plasma from Human
Subsets of miRNAs and non-coding RNAs are valuable disease biomarkers because of their cell-type specificity and abundance. We recently developed barcoded small RNA sequencing for miRNA profiling of large clinical sample collections from as little as 5 ng of total RNA starting material. Studying cell-free serum and plasma samples from trios of newborn babies and their parents we detected placental-specific miRNA in both maternal and newborn circulations and quantitated the relative contribution of placental miRNAs to the circulating pool of miRNAs. By introducing calibrator synthetic oligonucleotides during library preparation, we were able to calculate the total as well as specific concentrations of circulating miRNA. Furthermore, sequence variation in the placental miRNA profiles could be traced to the specific placenta of origin. In a separate study, through profiling and discriminant analysis of two skin tumors with shared histologic features, namely basal cell carcinoma (BCC) and Merkel cell carcinoma (MCC), we identified a distinct, inverse relationship between miR-205 and miR-375 expression that differentiated these tumors. We designed probes targeting these tumor-specific biomarkers and rRNA to establish multicolor miRNA fluorescence in situ hybridization (FISH) on formalin-fixed paraffin-embedded (FFPE) tissue sections. We quantified the fluorescence signals and validated our method on 16 BCC and MCC tumors, correctly identifying all tumors in a blinded analysis. These two examples serve as models for tumor or disease detection and document the usefulness of miRNA in molecular diagnostics and as circulating biomarkers. (Please see research articles for full list of authors. Clinical samples were obtained from various collaborations, and many additional laboratory members participated in technical and computational aspects of the studies).
Institute of Bioorganic Chemistry, Poznan, Poland
Finding RNA Structure in Influenza and Using Isoenergetic Microarrays to Reveal Oligonucleotide Binding Sites
Programs that predict the thermodynamic stability of RNA folding identified secondary structures in influenza A (1). Chemical and enzymatic mapping verified structures in mRNA (2). Oligonucleotide microarrays of 2′-O-methyl RNA pentamers and hexamers were designed to probe binding sites on several structures. Chemical modifications, including LNA and 2,6 diaminopurine, were included in the probes so that the stabilities of duplexes formed with unstructured RNA are relatively independent of base composition and sequence. The results suggest that binding to structured RNA is not easily predicted, but that microarray methods can rapidly suggest sequences that can be used in a chemical genetics approach to test for function. (1) W.N. Moss, S.F. Priore, and D.H. Turner (2011) Identification of conserved RNA secondary structure throughout influenza A coding regions, RNA 17, 991-1011. (2) W.N. Moss, L.I. Dela-Moss, E. Kierzek, R. Kierzek, S.F. Priore, and D.H. Turner (2012) The 3′ splice site of Influenza A segment 7 mRNA can exist in two conformations: A pseudoknot and a hairpin, PLoS ONE 7(6): e38323.
EllingtonAndy
University of Texas
Why Induced Fit Matters
One of the remarkable features of nucleic acids is that they can undergo facile conformational changes, and those conformational changes can be programmed to occur in response to ligands. The most obvious example is a molecular beacon. That said, aptamers have also been shown to undergo programmed conformational changes. However, there is a huge difference between a molecular beacon and an aptamer beacon, in terms of the thermodynamic and kinetic parameters that they obey, and also in terms of the ways in which they can be programmed. To a first approximation, a molecular beacon can undergo induced fit with its ligand, while an aptamer cannot. This fundamental difference has to a large extent been ignored when thinking about aptamers as biosensors and even as drugs. In order to better show the impact of induced fit conformational mechanisms, the role of both molecular beacons and aptamers in nucleic acid circuits will be highlighted.
WadaTakeshi
University of Tokyo
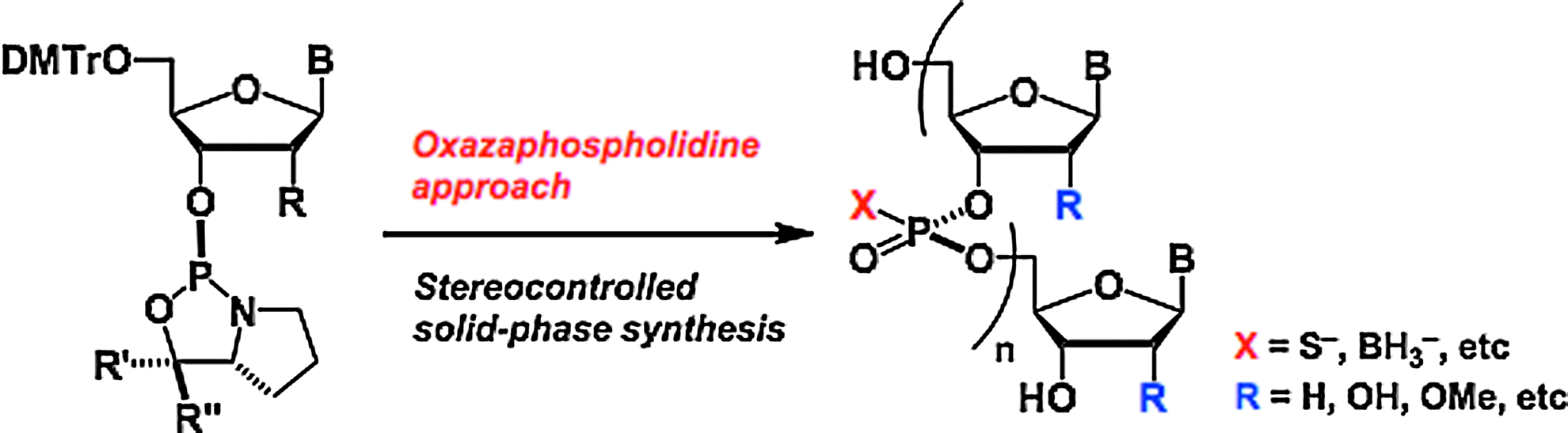
Stereocontrolled Synthesis and Properties of P-chiral Oligonucleotides
In recent years, a great deal of attention has been focused on small DNA and RNA molecules as therapeutic agents for selective inhibition of gene expression. Oligonucleotide therapeutic agents, such as antisense DNA, miRNA, and siRNA act on a target mRNA and arrest the protein synthesis. In order to stabilize these molecules in cells, a proper chemical modification of internucleotidic phosphodiester bonds is quite effective. Phosphorothioate DNA is one of the most widely used backbone-modified antisense DNAs. However, the currently used phosphorothioate DNAs are random mixtures of diastereomers since the chirality of the phosphorous atom cannot be controlled by use of the current synthetic methods. Because the properties of phosphorothioate DNAs, such as hybridization abilities with mRNA, affinities to proteins, and tolerances against nucleases are affected by the chirality of the phosphorous atoms, it is an important subject to develop an efficient method for obtaining stereoregulated phosphorothioate DNAs. Recently, we have developed a novel approach for the stereocontrolled synthesis of phosphorothioate DNAs and RNAs by the use of nucleoside oxazaphospholidine derivatives as monomers and N-(cyanomethyl)ammonium salts as activators (oxazaphospholidine approach). In this lecture, I wish to describe a recent progress of stereocontrolled synthesis of Pchiral DNA and RNA analogs, which are useful as oligonucleotide therapeutic agents, by the oxazaphospholidine approach.
SethPunit
Isis Pharmaceuticals
Structure-Activity Relationships of RNase H Antisense Oligonucleotides
Multiple classes of nucleic acid analogs which increase affinity for complementary RNA were profiled for their ability to increase the potency of second generation antisense oligonucleotides in animals. As part of this effort, the biophysical, structural and biological properties of oligonucleotides modified with 2′,4′-bridged nucleic acids (BNA or LNA), alpha-L-locked nucleic acids (alpha-L-LNA), hexitol nucleic acids (HNA), tricyclo DNA (tcDNA) and 2′-modified furanose analogs were characterized. Details from these profiling efforts which resulted in the selection of a new chemistry for use in human trials will be reported.
Over the past ten years, we have developed multiple conjugation strategies with the goal of robust in vivo delivery of siRNA for therapeutic use. Chemical modifications, the mode of administration, and the chemical nature of the conjugated ligands plays significant role in systemic delivery of siRNA. In early work, we demonstrated that covalent conjugation of small molecules such as cholesterol or other lipophiles to siRNA can enhance in vivo activity of RNAi therapeutics1–4. More recently, systemic delivery of siRNA to the liver has been achieved through conjugation of a multivalent N-acetylgalactosamine (GalNAc) ligand that targets the asialoglycoprotein receptor (ASGPR) expressed on the cell surface of hepatocytes. The ASGPR is highly expressed on liver hepatocytes and is highly conserved across species.5,6 ASGPR recognizes exposed terminal galactose (Gal) glycans and enables clearing of serum glycoproteins via clathrin-mediated endocytosis. Favorable recognition and binding by the ASGPR receptor requires multi-valency with appropriate spatial distribution and orientation of the Gal as well as GalNAc residues. Binding affinities of these ligands vary from mM to low nM and depend on the number of sugar residues present.7
We first synthesized a trivalent GalNAc building block attached to a tris scaffold and a hydroxyprolinol linker and used this to build one of the strands of the siRNA duplex (Figure 1).8 The trivalent GalNAc siRNA conjugates had nM binding affinities for the ASGPR, similar to trivalent GalNAc ligand reported in the literature.9,10 We have demonstrated liver-specific silencing of genes via subcutaneous administration of siRNA-GalNAc conjugates across multiple species (mouse, rat, and non-human primates (NHPs)). Subcutaneous administration of these conjugates resulted in high liver exposure and efficacy relative to the unconjugated siRNA with ED50s in the low mg/kg range. Furthermore, assessment of GalNAc-siRNA conjugate safety in rats and NHPs revealed a large safety margin. The GalNAc-siRNA conjugates hold significant promise as therapeutics targeting disease-causing genes expressed in liver.
SoutschekJ.et al.“Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs.”Nature, 2004; 432:173–178.a-36
KruetzfeldtJ.et al.“Silencing of microRNAs in vivo with ‘antagomirs’.”Nature, 2005; 438:685–689.a-37WolfrumC.et al.“Mechanisms and optimization of in vivo delivery of lipophilic siRNAs.”Nature Biotech., 2007; 25:1149–1157.a-38TomokoN.et al.“Harnessing a Physiologic Mechanism for siRNA Delivery with Mimetic Lipoprotein Particles.”Molecular Therapy 10.1038/mt.2012.33a-39ConnollyD.T., TownsendR.R., KawaguchiK., BellW.R., LeeY.D.J. Biol. Chem., 1982; 257:939–945.a-40AshwellG., MorellA.G.Adv. Enzymol., 1974; 41:99–128.a-41BiessenE.A.L., BeutingD.M., RoelenH.C.P.F., van de MarelG.A., Van BoomJ.H., van BerkelT.J.C.J. Med. Chem., 1995; 38:1538–1546.a-42Alnylam PharmaceuticalsUS8106022January12, 2012.a-43SevergniniM., ShermanJ., SehgalA., JayaprakashK. N., AubinJ., WangG., ZhangL., PengC. G., YuciusK., ButlerJ., FitzgeraldK.Cytotechnology, 2012; 64:187–195.a-44RensenP. C. N., SliedregtL. A. J. M., FernsM., KievietE., van RossenbergS. M. W., van LeeuwenS. H., van BerkelT. J. C., BiessenE. A. L. J. Biol. Chem., 2001; 276:37577–37584.a-45JadhavVasant
Merck
Optimization of Lipid Nanoparticles and siRNA Conjugates for Enablement of siRNA Therapeutics
The key to realize therapeutic potential of RNA interference (RNAi) is an effective and tissue specific delivery of short interfering RNA (siRNA). Unlike many small molecule drugs, the highly anionic and hydrophilic siRNAs are not readily taken up by cells to show target RNA silencing via RNA induced silencing complex (RISC) in cytosol. The current siRNA delivery approaches include encapsulation of siRNA in liposomes and conjugate technologies. Our efforts to improve these delivery vehicles will be presented.
Max Planck Institute of Molecular Cell Biology and Genetics, 01309 Dresden, Germany
AlCana Technologies, Vancouver, BC, V6L 2A1, Canada
Dept. of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC, Canada
Recent Advances in Lipid Nanoparticle-Mediated Delivery of RNAi Therapeutics
The advancement of RNAi therapeutics as a new class of innovative medicines has been enabled by the development of safe and efficient delivery platforms. Extensive structure-activity relationship (SAR) studies of the ionizable lipid component have led to the development of 2nd generation lipid nanoparticles (LNPs) with excellent in vivo efficacy. On the other hand, further mechanistic insights may facilitate rational optimization of this delivery platform. Here we report on studies aimed at dissecting the steps involved in tissue distribution, cellular uptake and intracellular trafficking of LNPs. Our time-resolved studies utilizing traceable siRNAs provide a unique insight into the kinetics of cellular uptake of siRNA-loaded LNPs in vitro and in vivo as well as their progression through the various cellular compartments. Functional delivery requires the release of siRNA from the endosomal compartment and loading into RISC. Based on EM analysis of cultured cells as well as liver sections from treated animals, we estimate that a low percentage of the siRNA escapes the endosomal pathway and reaches the cytosol. This is in good agreement with time course studies using stem-loop PCR for quantification of total and RISC-associated siRNA. Taken together, improved mechanistic insights and the design principles derived from lipid SAR studies provide a blueprint for a more rational approach to the development of LNPs with improved properties.
Axolabs GmbH
Roche Kulmbac
University of Illinois at Chicago
Dynamic PolyConjugate (DPC) Technology for Targeted Delivery of siRNA
We have developed a platform technology for systemic delivery of siRNA called Dynamic Polyconjugate (DPC). A key feature of DPC technology is a reversibly-masked, membrane-active polymer whose activity is revealed only in the acidic environment within endosomes. Masking also imparts pharmacokinetic properties to the DPC that are favorable for tissue-specific targeting. We have shown that attachment of the ligand N-acetyl galactosamine (NAG) directs DPCs specifically to liver hepatocytes in a receptor-dependent manner and where they are highly effective for target gene knockdown in rodents and non-human primates. Intravenous administration of DPCs containing 0.25 mg/kg siRNA in non-human primates results in >98% target gene knockdown after a single dose, with >80% knockdown for six weeks. Our latest generation DPC polymer is fully biodegradable, displaying rapid elimination kinetics while maintaining high siRNA delivery efficacy. Interestingly, we have found that simple co-injection of this masked, hepatocyte-targeted DPC polymer (NAG-polymer) and a liver-tropic, cholesterol-conjugated siRNA (chol-siRNA) results in highly effective target gene knockdown in both rodents and non-human primates at low chol-siRNA doses. This is accomplished without complex formation or interaction of the polymer and the chol-siRNA prior to reaching the target cell.
Currently, we are developing DPC technology for the treatment of chronic hepatitis B. According to the World Health Organization, some 360 million people, or 5% of the world's population, suffer from chronic hepatitis B and are at significant risk for development of liver cirrhosis and hepatocellular carcinoma. Existing drugs for the treatment of chronic hepatitis B seldom produce a functional cure and can cause significant side-effects. RNAi-based therapeutics have the potential to treat chronic hepatitis B virus infection in a fundamentally different manner than current therapies. Using RNAi, it is possible to knock down expression of viral RNAs, including the pregenomic RNA from which the replicative intermediates are derived, thus reducing viral load and the viral proteins that result in disease and impact the immune system's ability to eliminate the virus. We evaluated the efficacy of NAG-polymer to deliver chol-siRNAs in mouse models of chronic HBV infection. Co-injection of NAG-polymer with potent chol-siRNAs targeting conserved human HBV sequences resulted in profound repression of viral RNA, proteins and viral DNA with long duration of effect. Single injections in a replication-competent, transiently transgenic HBV mouse model resulted in greatly reduced levels of HBsAg (>3 log10), HBeAg (LOD) and HBV DNA (2–3 log10) in serum and HBV RNA in liver. HBsAg was reduced more than >99% for one month following a single injection. Serum HBeAg and HBV RNA, DNA and core antigen in liver were also dramatically reduced in the transgenic HBV1.3.32 mouse model of chronic HBV infection. These results suggest that co-injection of the hepatocyte-targeted, endosomolytic NAG-polymer and chol-siHBVs holds great promise as a new therapeutic for patients chronically infected with HBV.
BartelDavid
HHMI/MIT/Whitehead Institute
microRNAs and their Regulatory Effects
We have been using molecular and computational approaches to find regulatory RNAs, identify the messages that they regulate, and then investigate molecular consequences of these regulatory interactions and their functions during development, oncogenesis, and other biological processes. This talk will describe our current understanding of microRNAs. Potential topics include how the cell distinguishes microRNA precursor transcripts from all the other transcribed RNAs, how microRNAs recognize their targets, the identities of the regulatory targets, and the extent to which microRNA-directed repression can be explained by destabilization of the targeted mRNAs.
SarnowPeterMachlinEricaSaganSelena
Stanford University
Protection of the hepatitis C viral RNA genome and modulation of polyadenylation site usage in Insig1 mRNA by liver-specific pre- and mature microRNA 122
In general, microRNAs interact with sites residing in 3' noncoding regions in target mRNAs, leading to posttranscriptional downregulation of mRNA expression. In contrast, liver-specific microRNA miR-122 is known to bind at two adjacent sites that are close to the 5' end of the hepatitis C virus (HCV) RNA genome, resulting in upregulation of viral RNA abundance. Results obtained with mutated HCV genomes argue that both sites are likely to be occupied in the same molecule for efficient regulation to occur. We discovered that the location of the miR-122 binding site in the viral genome dictates it's effect on gene regulation, because insertion of the miR-122 binding site into the 3' noncoding region of a reporter mRNA leads to downregulation of mRNA expression, suggesting that miR-122 can interact with RNA targets via a typical RISC complex. Curiously, defined mutations at distinct locations in miR-122 abolished upregulation of viral RNA, yet functioned well in microRNA-dependent downregulation of reporter mRNA expression. This finding suggests that the HCV-miR-122 complex is distinct from RISC-miR-122 complexes that regulate cellular target mRNAs in the liver. Extensive analyses have shown that miR-122 regulates the turnover of HCV RNA, with only minimal effects on viral mRNA translation or rates of replication. To explore the feasibility of a novel antiviral intervention by targeting an oligomeric microRNA-HCV complex, it is important to understand the roles for miR-122 in the liver. Several studies have shown that sequestration of miR-122 by antisense-oligonucleotides in the livers of mice and non-human primates lowered the abundance of cholesterol and downregulated fatty acid biosynthesis. We have discovered that a major target for pre-miR-122 is Insig1 mRNA. Insig 1 functions as an inhibitor of liver-specific transcription factor SREBP that modulates the transcription of many genes in the cholesterol biosynthesis pathway. Specifically, pre-miR-122 downregulates the synthesis of an Insig1 mRNA isoform that is used for Insig1 protein synthesis. Thus, sequestration of pre-miR-122 offers an attractive strategy for the temporal downregulation of HCV and cholesterol in infected livers.
MooreKathryn
New York University School of Medicine
miR-33: A Therapeutic Target for Cardiometabolic Diseases
MicroRNAs have emerged as important post-transcriptional regulators of lipid metabolism, and represent a new class of targets for therapeutic intervention. MicroRNA-33a and b (miR-33a/b) are intronic microRNAs embedded in the sterol response element binding protein genes SREBF2 and SREBF1, which upon co-transcription, cooperate with their host genes to balance cellular lipid levels. Work from our group and others identified the cholesterol transporter ABCA1 - a key regulator of HDL, “the good cholesterol” - as one of the major targets of the miR-33 family. In studies performed in mice, we established that inhibiting miR-33 (miR-33a in humans) is an effective strategy to increase plasma HDL and reverse cholesterol transport, and to regress atherosclerosis. Although highly promising, the translational insight of these studies was limited by the fact that mice lack miR-33b, which is present in larger mammals and may contribute substantially to total miR-33 levels in humans. Thus, to gain a comprehensive understanding of the effects of inhibiting both isoforms of miR-33 in a clinically-related model, we treated African green monkeys with an antisense miR-33 oligonucleotide, that effectively inhibits both miR-33a and miR-33b. Systemic delivery of anti-miR33 over 12 weeks increased hepatic expression of ABCA1 and induced a sustained 50% increase in plasma HDL. Notably, miR-33a/b antagonism also increased the hepatic expression of additional miR-33a/b target genes involved in fatty acid oxidation and, unexpectedly reduced genes involved in fatty acid synthesis. These additional effects resulted in a marked suppression of plasma levels of VLDL-associated apolipoprotein B and triglycerides (both risk factors of cardiovascular disease) not previously detected in mice. These data establish, in a model highly relevant to humans, that pharmacological inhibition of miR-33a/b is a promising therapeutic strategy to raise plasma HDL and lower VLDL triglycerides for the treatment of dyslipidemias that increase cardiovascular disease.
St. Louis University School of Medicine
Targeting alpha1-antitrypsin for the Treatment of A1AT Liver Disease
Alpha-1 antitrypsin deficiency (AATD) is a rare genetic disease due to mutations of the alpha-1 antitrypsin (A1AT) gene. Emphysema associated with AATD is caused by the deficiency of systemic A1AT protein (loss-of-function phenotype) and can be treated with augmentation therapy. In contrast, AATD Liver Disease, which is caused by aggregation and retention of mutant A1AT protein in the liver, can only be treated by liver transplantation. The hepatic pathology is the result of a toxic gain-of-function of the mutant Z protein, which affects both children and adults. We have developed optimized 2′-O-methoxyethyl (“2nd Generation”) phosphorothioate antisense oligonucleotide (ASO) drugs targeted against A1AT to determine if reducing the expression of A1AT production in liver would improve AATD-related liver disease. ASOs were designed to utilize an RNaseH-dependent mechanism of action, which promotes cleavage of A1AT mRNA. Administration of A1AT ASOs to PIZ transgenic mice expressing the mutant human gene resulted in significant reductions in hepatic A1AT mRNA and circulating A1AT levels that were dose dependent. Importantly, ASO treatment of PIZ mice produced significant reductions in A1AT protein aggregates in liver, reduced markers of fibrosis (e.g., TIMP1), and slowed fibrosis progression. Futhermore, administration of A1AT ASOs in non-human primates (50 mg/kg×12 wk) resulted in marked reductions in hepatic and circulating A1AT mRNA and protein levels. Our data demonstrate that antisense oligonucleotide treatment can potently reduce mutant A1AT protein production and represents a potential treatment for AATD liver disease.
RNAi-Mediated Inhibition of a Natural Anticoagulant for the Treatment of Hemophilia
Significant progress has been made in the treatment of hemophilia since the advent of recombinant replacement factor therapy. Nonetheless, short half-life, cost, complications associated with repeated vascular access, and in particular, the development of inhibitors to replacement factor, leave significant unmet need for new therapeutic approaches. In hemophilia, the loss of procoagulant factors (Factor VIII (FVIII) and Factor IX (FIX), in the case of hemophilia A and B, respectively) results in an insufficient generation of thrombin, the key enzyme converting fibrinogen to insoluble fibrin. This leads to an imbalance of the hemostatic system toward a bleeding phenotype. Interestingly, there have been reports suggesting that coinheritance of prothrombotic mutations (e.g. Factor V Leiden, protein C deficiency, protein S deficiency, antithrombin deficiency, prothrombin G20210A) may ameliorate the clinical phenotype in hemophilia. We are currently investigating the use of RNA interference (RNAi) to target the natural anticoagulant antithrombin (AT) as strategy to rebalance the hemostatic system and improve thrombin generation in hemophilia. We have developed a novel, chemically-modified, ligand-conjugated short interfering RNA (siRNA), ALN-AT3, against AT. A trivalent N-acetylgalactosamine ligand was utilized to achieve targeting to hepatocytes, the site of AT synthesis and secretion. ALN-AT3 demonstrates potent activity in mice after single subcutaneous (SC) administration (ED50 ∼ 1 mg/kg). The PK properties of this siRNA have been characterized in mice, indicating good bioavailability and liver exposure. ALN-AT3 treatment results in persistent AT reduction, suggesting a weekly or twice-monthly SC dosing paradigm. Dose regimen studies in mice have confirmed the feasibility of these dosing regimens, demonstrating sustained 80% reduction of AT at a weekly dose of 0.75 mg/kg. Further, treatment of hemophilia mice with ALN-AT3 has resulted in normalization of thrombin generation. Translation of ALN-AT3 activity in higher species has recently been achieved. A single SC administration of ALN-AT3 in nonhuman primates resulted in dose-dependent reductions of plasma AT of 50%, 70%, and 80% at 1, 3, and 10 mg/kg, respectively. These data suggest that the use of a novel RNAi therapeutic targeting AT is a promising approach for the treatment of hemophilia, and potentially, other bleeding disorders. Further, the SC route of administration, long duration of action, and applicability to persons with hemophilia who have inhibitors, make this a particularly encouraging potential therapy.
MirkinChad
Northwestern University
Spherical Nucleic Acids: Novel Topical Agents for the Treatment of Skin Disease and Brain Cancer
The natural defenses of biological systems for exogenous oligonucleotides, such as synthetic antisense DNA and siRNA, present many challenges for the delivery of nucleic acids in an efficient, non-toxic, and non-immunogenic fashion. Because nucleic acids are negatively charged and prone to enzymatic degradation, researchers have relied on transfection agents such as cationic polymers, liposomes, and modified viruses to facilitate cellular entry and protect delivered biomolecules. However, each of these platforms is subject to several drawbacks, including toxicity at high concentrations, the requirement of specialty nucleic acids to enhance stability, and severe immunogenicity.
Spherical nucleic acid (SNA) gold nanoparticle conjugates (inorganic nanoparticle cores functionalized with a spherical shell of densely organized, highly oriented nucleic acids) pose one possible solution to these problems in the context of antisense and RNAi pathways. Remarkably, these highly negatively charged SNA structures do not require cationic transfection agents or additional particle surface modifications and naturally enter all cell lines tested to date (over 50, including primary cells). Further work has shown the cellular uptake of these particles to be dependent upon DNA surface density: higher densities lead to higher levels of particle uptake. The high-density polyvalent nucleic acid surface layer is believed to recruit scavenger receptors from the cells that facilitate endocytosis. Moreover, the ion cloud associated with the high-density oligonucleotide shell, combined with steric inhibition at the surface of the particles, inhibits enzymatic nucleic acid degradation and activation of the enzymes that trigger the innate immune response of certain cells. Methods to synthesize such structures and current cellular and animal work focused on developing them as single-entity agents for the treatment of skin disease and brain cancer will be described. These new methodologies take advantage of the interesting properties unique to spherical and other forms of three-dimensional nucleic acids.
SlackFrank
Yale University
MicroRNAs as Targeted Therapeutics and Therapeutic Targets in Cancer
MicroRNAs are small non-coding RNAs that regulate gene expression to control important aspects of development and metabolism such as cell differentiation, apoptosis and lifespan. let-7 encodes a microRNA implicated in human cancer. Specifically, human let-7 is poorly expressed or deleted in lung cancer, and over-expression of let-7 in lung cancer cells inhibits their growth, demonstrating a role for let-7 as a tumor suppressor in lung tissue. let-7 is expressed in the developing mammalian lung and regulates the expression of important oncogenes implicated in lung cancer, suggesting a mechanism for let-7's involvement in cancer. We are focused on the role of let-7 and other oncomiRs in regulating proto-oncogene expression during development and cancer, and on using miRNAs to suppress tumorigenesis.
UhlmannEugen
AdiuTide Pharmaceuticals
Pre-Clinical Development of Novel Immune Stimulatory CpG Oligonucleotides With Improved Recognition and Activation of TLR9
The Ohio State Medical Center
Center for Neuromuscular and Neurological Disorders, University of Western Australia
Department of Gene Therapy Nationwide Children Hospital
Treatment of Spinal Muscular Atrophy with Antisense Morpholinos that Alter Splicing of SMN2 Good Distribution in Neonates and Adult Animals
Spinal Muscular Atrophy (SMA) is an autosomal recessive disorder and the leading genetic cause of infant mortality. The disorder is characterized by motor neuron loss in the anterior horn of the spinal cord. SMA is caused by loss or mutation of the Survival Motor Neuron 1 gene (SMN1) and retention of the SMN2 gene. The SMN2 gene copy number varies in the human population and the severity of the SMA phenotype is inversely related to the SMN2 copy number. The SMN1 and SMN2 genes differ essentially by a single nucleotide lying in exon7 which disrupts a splice modulator and results in SMN2 producing mostly transcript lacking exon 7. Within both intron 6 and intron 7 there are numerous sequences important for the regulation of SMN exon 7 incorporation both in a positive and negative manner. Of particular interest are the negative regulators of SMN exon 7 incorporation which as a general rule bind the negative splice regulator hnRNPA1. We have used a morpholino based antisense oligonucleotide (ASO) to block this sequence and tested this treatment in SMA mice. The SMA mice used possess two copies of human SMN2, a transgene expressing SMN lacking exon 7 (SMND7) and lack mouse Smn (SMN2+/+; SMNΔ7; Smn−/−). These mice usually live 13–14 days. A single intracerebroventricular (ICV) injection of morpholino ASO at day 1 resulted in an extension of life to over 100 days latter injections also gave improvement but to a reduced extent. The incorporation of SMN exon7 showed a clear dose response curve to amount of ASO and SMN levels increased in all cases. With time the amount of SMN as well as the amount of exon7 included by SMN2 decreased. However both transcript and SMN protein remained elevated at 65 days. There was no evidence for a peripheral role for high SMN levels in these mice. A re-dosing of SMA mice at 30days with bare morpholino delivered by ICV in a single bolus sterotactic injection did not increase survival of SMA mice. However subsequent analysis revealed that the bare morpholino did not spread in the adult spinal cord. We explored two methods to increase the spread of morpholino throughout the nervous system in the first we annealed the antisense morpholino with a sense DNA strand, in the second we mixed the morpholino with a specific reagent (reagent1). The addition of charge with the DNA did increase distribution and uptake of the morpholino ASO. However reagent 1 is simpler and results in even greater distribution of the ASO furthermore this is applicable in a clinical situation. We are now exploring administration of morpholino at day one followed by delivery of the morpholino at day 30 in reagent 1 using osmotic pumps delivering the ASO over a month to determine if this further increases survival of SMA animals. Furthermore we are exploring the use of reagent 1 complexed morpholinos for efficient delivery throughout the nervous system of the neonatal pig.
University of Rochester Medical Center, Rochester, NY
Isis Pharmaceuticals, Carlsbad, CA
Genzyme Corporation, Framingham, MA
Correction of myotonic dystrophy in mouse models by antisense targeting of nuclear-retained RNA
Myotonic dystrophy type 1 (DM1) is one of several dominantly-inherited disorders in which genomic expansions of tandem repeats lead to expression of repetitive RNAs that have deleterious effects. This RNA gain-of-function mechanism is implicated in both types of DM, several forms of cerebellar degeneration, and the most common form of familial ALS. In DM1 the repetitive segment is an expanded CUG repeat (CUGexp) in the 3' untranslated region of DM protein kinase (DMPK) mRNA. The mutant RNA accumulates in nuclear foci, trapping proteins that have poly(CUG) binding affinity. This leads to misregulated alternative splicing and other perturbations of the muscle transcriptome. Antisense drugs would seem ideally suited for treating RNA toxicity, were it not for the problem of low biodistribution to muscle. This presentation will focus on evidence that nuclear-retained transcripts are unusually sensitve to RNase H-active antisense oligonucleotides (ASOs), enabling therapeutic knockdown in skeletal muscle. In transgenic mouse models of DM1, the systemic administration of 2′-0-methoxyethyl gapmer ASOs produced >80% knockdown of CUGexp RNA in skeletal muscle. Strong knockdown was achieved within 4 weeks (8 subcutaneous injections) and persisted for one year after cessation of treatment. No specific changes of ASO design or formulation were required, and the effect did not depend on enhanced entry of ASOs into muscle fibers. Many of the physiological, morphological, and biochemical features of the disease were corrected, without evidence for toxicity or off-target knockdown. These results suggest that the ASO-RNase H mechanism may provide an effective strategy for treating RNA dominant diseases.
Program in Cellular and Molecular Medicine, Boston Children's Hospital, Harvard Medical School
Massachusetts General Hospital
Ragon Institute of MGH, MIT & Harvard
Knocking Down HIV Transmission Using CD4 Aptamer-siRNAs
The continued spread of the HIV epidemic underscores the need to interrupt transmission. One attractive strategy is a topical vaginal microbicide. Sexual transmission of herpesvirus type 2 (HSV-2) in mice can be inhibited by intravaginal siRNA application. Gene knockdown in the mouse genital tract and protection from HSV transmission is durable, lasting weeks, suggesting that a microbicide based on siRNAs as the active ingredient might not need to be applied just before sexual intercourse, when compliance is problematic. However, knocking down gene expression in immune cells susceptible to HIV infection is challenging. Chimeric RNAs composed of an aptamer fused to an siRNA can be used for targeted gene knockdown in cells bearing an aptamer-binding receptor. We used a previously identified aptamer that recognizes human CD4, the receptor for HIV entry into cells, to design CD4 aptamer-siRNA chimeras (CD4-AsiCs) that are taken up specifically by CD4+ lymphocytes, monocytes and macrophages, processed by Dicer within cells and induce gene knockdown. CD4-AsiCs specifically suppress gene expression in CD4+ T cells and macrophages in polarized cervicovaginal tissue explants and in the female genital tract of immunodeficient mice transplanted with human hematopoietic stem cells and fetal thymus (“humanized mice”). CD4-AsiCs do not activate lymphocytes or stimulate innate immunity. CD4-AsiCs that knockdown HIV genes and/or the HIV coreceptor CCR5 inhibit HIV infection in vitro and in tissue explants. When applied intravaginally to humanized mice, CD4-AsiCs protect against HIV vaginal transmission. A cocktail combining siRNAs against CCR5 and viral genes is more effective than CCR5 or antiviral RNAs individually. Gene silencing lasts for ∼15 d. However, mice challenged a week after CD4-AsiC administration are only partially protected against viral challenge. Thus CD4-AsiCs could be used as the active ingredient of a microbicide to prevent HIV sexual transmission. In preliminary studies, we also have been able to use CD4-AsiCs to induce gene knockdown in CD4+ cells systemically in the spleen and distal lymph nodes.
Santaris a/s Pharma, DK
Development of Miravirsen an Oligonucleotide Directed Against miR-122 : Clinical Study Results
Hepatitis C virus (HCV) is a major cause of chronic liver disease, cirrhosis, and hepatocellular carcinoma and is the leading indication for liver transplantation in the United States and Europe. The World Health Organization (WHO) estimates that 180 million people (3% of the World's population) are infected with HCV. Miravirsen sodium is a β-D-oxy-Locked Nucleic Acid (LNA) modified phosphorothioate anti-sense oligonucleotide inhibitor of the liver-expressed microRNA-122 (miR-122). miR-122 binds to two closely spaced miR-122 target sites (S1 and S2) in the 5′ untranslated region (UTR) of the HCV genome, and forms an oligomeric miR-122-HCV complex, thereby protecting the 5′ HCV genome from nucleolytic degradation (Jopling et al., Science 2005; Machlin et al., PNAS 2011; Shimakami et al. PNAS 2012). Thus, miravirsen is a host targeting agent that reduces HCV RNA levels indirectly, by targeting the critical host factor miR-122, rather than by directly targeting the virus. In clinical trials in healthy volunteers, miravirsen was safe and well tolerated when given by weekly subcutaneous injection over 29 days (five injections) at doses up to 5 mg/kg. No dose limiting toxicities were identified. In a four-week monotherapy trial, SPC3649-203, conducted in genotype 1 subjects with chronic HCV infection, the mean reduction in HCV RNA for the 3, 5 and 7 mg/kg dose groups (five weekly doses over 29 days) was approximately 1, 2 and 3 log10, respectively. Decreases were first noticeable after 3 doses of miravirsen, with maximal mean declines in HCV RNA observed 4 to 8 weeks after the end of treatment. The observed decline in HCV RNA was sustained well after the last dose of miravirsen. The reduction in group mean HCV RNA levels was correlated to miravirsen dose level and group mean plasma exposure (trough and peak). There was no evidence of genotypic resistance.
Over ten years ago, Elbashir and colleagues (Nature 411(6836):494-8, 2001) described the use of small interfering RNAs (siRNAs) as mediators of RNAi in a range of mammalian cell lines. However, despite numerous publications since then demonstrating in vivo RNAi in a variety of species, unequivocal proof of siRNAs as pharmacological mediators of gene silencing in man have been lacking. The major obstacle towards this goal has been safe and effective delivery of RNAi therapeutics to target organ(s). We have made significant progress recently using lipid nanoparticles (LNP) to effect hepatic siRNA delivery. Importantly, these efforts have enabled clinical transition and POC studies for multiple programs. We will present preliminary data from three Phase 1 studies; ALN-TTR01 and -TTR02, for TTR-mediated amyloidosis; and ALN-PCS for hypercholesterolemia, showing that RNAi can be used in humans as a safe and effective method against important disease-causing transcripts. In both programs, the use of circulating protein biomarkers shows that potent, dose-dependent, and durable RNAi is observed after a single intravenous dose of an LNP-encapsulated siRNA, and that the pharmacologic profile closely resembles that previously described in nonhuman primates.
MoniaBrett
Isis Pharmaceuticals
Development of Antisense Drugs for Cancer
Oncology drug discovery and development is rapidly moving towards a “Personalized Medicine” approach in which cancer therapy is tailored to the genetic or epigenetic cause of an individual's cancer. The foundation for this approach is genomic sequencing of cancer cells to identify causal mutations, which facilitates both choice of treatment and the identification of novel targets for cancer drug discovery and development. Antisense technology is particularly well suited to exploit this new paradigm for cancer drug discovery due to its ability to rapidly identify drugs against all targets regardless of their function, and to impact target function with unprecedented specificity. However, antisense drug development for cancer has historically had mixed results, and the need for greater clinical success is high. Research into the discovery of new antisense chemistries that are more effective in penetrating and producing robust antisense effects in cancer cells is progressing rapidly, and resulting in more reproducible and robust effects in preclinical cancer models and in the clinic. We have been developing a bicyclic nucleic acid chemistry, referred to as constrained ethyl (cET), for a number of disease indications including cancer. The most advanced cET antisense program is the ISIS-STAT3rx program for cancer. STAT3 is a transcription factor that resides within the JAK-STAT signaling pathway, and mutations leading to the chronic activiation of STAT3 have been strongly linked to a variety of hematological and solid cancers. ISIS-STAT3rx potently and reproducibly lowers levels of STAT3 in cell culture, and in tumor cells and tumor-associated stromal cells in preclinical models, and produces robust antitumor activity in STAT3-dependent tumor models. ISIS-STAT3rx is currently under evaluation in a Phase I study in patients with cancer and early results are encouraging. Results will be presented demonstrating the utility of the cET antisense approach for cancer including a review of the ISIS-STAT3rx program.
MüllerPhilipp
Cytos Biotechnology
Efficacy of CYT003-QbG10 - an A-type CpG Delivered in Virus-like Particles - in Patients with Allergic Asthma
CYT003-QbG10 (QbG10) is a virus-like particle (VLP Qb), containing an A-type CpG (G10). We postulate efficient uptake by pDCs after s.c. injection, targeted delivery to the lymph node, release of the TLR9 agonist and induction of Th1 cytokines. Phase II trials in patients with rhinoconjunctivitis showed improved allergic symptoms compared to placebo. The present study was a multi-center, parallel-group, double-blind, randomized and placebo controlled Phase IIa clinical trial in 63 patients with persistent allergic asthma that required long term treatment with inhaled corticosteroids (ICS). Patients were randomized to 7 s.c. injections of placebo or 0.9mg QbG10 at weekly/bi-weekly intervals. After a stable add-on phase, the ICS dose was reduced after 4 weeks to 50% and completely withdrawn after 8 weeks, if possible. Clinical efficacy was assessed over the 12 week study period by the Juniper Asthma Control Questionnaire (ACQ), by electronic diaries in which daytime and nighttime asthma symptoms and use of relief-medication were recorded, and by spirometry (FEV1).
Results: All objective and patient reported outcome parameters were significantly improved versus placebo from week 6 onward to the end of the study. The fraction of patients whose allergic asthma was “well controlled” (ACQ score ≤0.75) increased from 42% under ICS therapy to 67% under QbG10 treatment despite corticosteroid withdrawal, while under placebo the fraction of “well controlled” patients fell from 40% to 33% (p=0.008). The average ACQ score was significantly improved versus placebo from week 4 until week 12 and remained below 0.75 points under QbG10 treatment in all but one week. Under placebo treatment, the ACQ score gradually deteriorated and approached the threshold for "loss of asthma control" (1.5points) at the end of the study. Furthermore, daytime and nighttime asthma symptoms, use of relief medication as well as lung function (FEV1) were all significantly improved vs. placebo. Treatment was safe and generally well tolerated, with local reactions being the most frequent events.
Discussion: Achieving asthma control is an important therapeutic goal. CYT003-QbG10 has the potential to provide patients with a therapeutic alternative to either replace or complement commonly used asthma medications, e.g. inhaled corticosteroids, while maintaining or improving asthma control. The current study demonstrated that CYT003-QbG10 improved asthma control, lowered symptoms and use of relief medication and improved lung function versus placebo in an ICS withdrawal setting. Further trials in patients with uncontrolled asthma despite use of ICS and other controller therapies at recommended doses (GINA steps 3+4) will be undertaken.
KayeEd
Sarepta Therapeutics
Results of the Eteplirsen Phase 2b and Phase 2b Extension Study in Duchenne Muscular Dystrophy
Introduction: DMD is a rare, degenerative, genetic disease that results in progressive muscle loss and premature death. Affecting 1 in 3500 male births, DMD is caused by the inability to produce the dystrophin protein. There are currently no approved drugs or disease-modifying therapies available to treat DMD. Eteplirsen is an investigational therapy designed to enable functional dystrophin production in boys who are amenable to exon 51-skipping therapy (∼13%).
Methods: This Phase 2b study is an open-label extension evaluating eteplirsen in patients who completed the 24 week double-blind, placebo-controlled phase of the study. Twelve patients were enrolled across three cohorts: 4 received 50mg/kg of eterplirsen weekly for 48 weeks, 4 received 30mg/kg of eteplirsen weekly for 48 weeks, and 4 received placebo for 24 weeks, followed by eteplirsen for 24 weeks. The primary endpoint is the change in the percent of dystrophin positive fibers from baseline and the primary clinical outcome measure is the change in the 6-minute walk test (6MWT) distance from baseline, both compared to the placebo/delayed treatment cohort.
Results: Results from the double-blind placebo controlled portion of the Phase 2b study demonstrated a statistically significant (p≤0.002) increase in novel dystrophin (22.5% dystrophin-positive fibers as a percentage of normal) in the group treated with eteplirsen for 24 weeks (30mg/kg weekly), compared to no increase in the placebo group,. Furthermore, a significant clinical benefit of 69 meters was observed (p<0.019) on the 6MWT through 36 weeks in the eteplirsen 50mg/kg weekly cohort compared to patients who received placebo for 24 weeks followed by 12 weeks of treatment with eteplirsen. No treatment-related adverse events were seen through 36 weeks.
Conclusions: Through 36 weeks of treatment, eteplirsen has demonstrated significant levels of novel dystrophin, a significant clinical benefit on 6MWT, and no treatment related adverse events. Data on these outcomes will be reported through 48 weeks of treatment.
RöhlIngo
Axolabs
Bioanalytical Tools for Optimization of siRNA Chemistry and Improved in vivo Functionality
A new hybridization based HPLC based assay for bio-analysis of therapeutic short interfering RNA (siRNA) will be presented. The assay system enables the separation and quantitation of the oligonucleotide parent compounds and their metabolites with a LOD lower than 1 ng/g from tissue and plasma. Assay based quantitative detection of the 5′-phosphorylated antisense strand serves as marker for cytoplasmic delivery of the siRNA. Monitoring the 5′-phosphorylated antisense strand as the in vivo activated drug substance is exploited for the establishment of a PK/PD relationship. Analyzing the kinetics of 5′-phosphorylation together with the characterization of metabolites provides to identify delivery system specific modification strategies to maximize siRNA activity in vivo.
JensenJeff J.
Inflammation and Remodeling Research Unit, Pfizer Worldwide R&D, La Jolla, CA
Development of a 2′-MOE Antisense Oligonucleotide as a Novel Approach to Treat Skin Scarring & Fibrosis
Connective tissue growth factor (CTGF) plays a fundamental role in scar formation, epithelial to mesenchymal transition and acting as a cofactor with TGFβ to induce and sustain fibrosis. In normal skin of adults, CTGF is undetectable but is strongly upregulated during injury, wound healing and fibrosis. PF-06473871 is a second generation antisense oligonucleotide (ASO) that targets CTGF and is under development for the treatment of dermal scarring. PF-06473871 was taken into clinical development by Excaliard Pharmaceuticals, which was acquired by Pfizer in 2011.
The blockade of CTGF pathways using ASOs has been shown to impact hypertrophic scarring in pre-clinical models, demonstrating both a reduction in CTGF expression as well as a reduction in indices of hypertrophic scarring. In Phase 2 clinical studies in healthy subjects, similar data have been generated using PF-06473871, demonstrating the effectiveness of the ASO approach in reducing mRNA and protein for CTGF as well as the ability to reduce the severity of skin scars in an experimental setting. Clinical proof-of-principle has been established in subjects having scar revision surgery of bilateral, hypertrophic breast scars. In this study, the reduction in scar severity was demonstrated following administration of PF-06473871, with efficacy shown both at 12 and 24 weeks post surgery.
The presentation will review the pre-clinical and clinical data generated to date.
GrundyJohn
Isis Pharmaceuticals
Pharmacokinetic and Pharmacodynamic Properties of Antisense Oligonucleotides: Bridging Nonclinical to Clinical
An understanding of the potential roles of RNAs in maintaining normal health and their contribution to various diseases has taken dramatic advances over the past decade and is only continuing to increase, such that the approach of directly targeting RNAs [with drugs such as second generation antisense oligonucleotides (ASOs)] presents a very compelling therapeutic strategy. The rapid development of antisense technology and the advancement provided by the second generation ASOs (as well as any other next generation ASO chemistries) offers an almost unlimited scope for the development of new and highly specific therapeutics with desirable pharmacokinetic, pharmacological, and safety properties. Multiple ‘second generation’ gapmer antisense oligonucleotides (ASOs), such as 2'-O-(2-methoxyethyl) partially modified (chimeric) compounds with fully phosphorothiated backbones, have been evaluated as potential therapeutic agents in the clinic. Compared to first generation chemistries, second generation ASOs consistently demonstrate greater biological stability, greater in vitro and in vivo potency, and typically fewer non-hybridization based toxicities. The general similarity of preclinical and clinical PK properties across the class of second generation ASOs, ease of formulation and parenteral administration (including but not limited to IV and SC routes), good SC systemic bioavailability, predictable distribution and post-distribution phase plasma kinetics, low to moderate intersubject/interanimal variability in PK exposure measures, long tissue half lives allowing for infrequent dosing, extensive metabolism to shortmers by ubiquitously expressed nucleases found within all cells followed by rapid renal elimination, and little or no potential for metabolic based (or protein binding based) drug-drug interactions, all help to facilitate rapid preclinical and clinical drug development with this class of compounds. For scaling of PK/PD properties from animals to humans and prediction of clinical pharmacologically active doses, five basic principles typically apply: 1) pharmacologic effects of ASOs are directly related to drug concentrations in the target organ [and appear to typically translate well from transgenic (Tg) mice to humans]; 2) ASO post-distribution plasma concentrations (e.g., trough levels) can provide a surrogate measure for estimating corresponding target tissue concentrations (particularly useful for the clinic); 3) monkey plasma PK exposure measures scale directly to humans based on comparable mg/kg dosing; 4) ASO tissue half-lives are long (typically 2–4 weeks) and appear very similar across species; and 5) initial clinical dosing is based on achieving steady-state trough plasma concentrations (and corresponding target tissue concentrations) consistent with those supporting PD activity seen in nonclinical models (preferably Tg mice). When the target organ is the liver, application of these principles typically estimates human pharmacologically active doses for second generation ASOs ranging from 50–400 mg/week and which have proven to be remarkably accurate. In this presentation, further illustration of these principles and actual clinical examples will be shown.
YounisHusamHenryScott P.
Isis Pharmaceuticals
Renal Tolerability of 2'-MOE Antisense Oligonucleotides (ASOs) and The Role of Drug Accumulation
Antisense oligonucleotides (ASO) distribute well to several tissues, and drug concentrations are typically greatest in the liver and kidney following systemic delivery. The relatively long tissue residence of ASO therapeutics is due to their long half-life (e.g. 2 to 4 weeks in liver or kidney), and affords infrequent dose administration (i.e. weekly to monthly). At relatively high doses drug accumulation of ASO therapeutics in the liver and kidney has been observed in animals. The objective of this presentation will review the safety profile of ASO therapeutics as it relates to drug accumulation in the kidney. The renal safety of the 2'-methoxy-ethyl (2'-MOE) class of ASOs will be described in rodents and non-human primates. The translation of findings observed in the kidney in animals to human safety will also be discussed.
miRagen Therapeutics
Hepatic Safety Across Species
Hepatotoxicity is a so-called « class effect » of antisense oligonucleotides (ASO). The main mechanistic hypothesis is non-covalent binding to intracellular proteins. This toxicity occurs at differing dose levels and over a wide dose range across compounds, enabling the selection of drug candidates with acceptable therapeutic indices. With a focus on Locked Nucleic Acid (LNA) compounds, and based on a small series of clinical LNA candidates, the predictive value of animal models is discussed, using conventional Drug-Induced Liver Injury (DILI) criteria.
PFRED – An Open Source Olignucleotide Design and Analysis Tool
Software for the design, analysis and visualization of antisense and siRNA oligonucleotides will be described. The code has been made available as an OpenSource project, which can be used and developed throughout the academic and industrial oligonucleotide community. Standardized workflows are included to help users identify the target sequence of interest from an EnsEMBL ID and apply appropriate design criteria for the selection of compounds for synthesis. Additionally, a new open heirarchacal notation language (HELM) for oligonucleotides and bioconjugates will be discussed. A public version of the server components will be hosted in the Amazon cloud computing environment.