
Abstract
Hereditary angioedema (HAE) is a rare disorder characterized by recurrent, acute, and painful episodes of swelling involving multiple tissues. Deficiency or malfunction of the serine protease inhibitor C1 esterase inhibitor (C1-INH) results in HAE types 1 and 2, respectively, whereas mutations in coagulation factor 12 (f12) have been associated with HAE type 3. C1-INH is the primary inhibitor of multiple plasma cascade pathways known to be altered in HAE patients, including the complement, fibrinolytic, coagulation, and kinin-kallikrein pathways. We have selectively inhibited several components of both the kinin-kallikrein system and the coagulation cascades with potent and selective antisense oligonucleotides (ASOs) to investigate their relative contributions to vascular permeability. We have also developed ASO inhibitors of C1-INH and characterized their effects on vascular permeability in mice as an inducible model of HAE. Our studies demonstrate that ASO-mediated reduction in C1-INH plasma levels results in increased vascular permeability and that inhibition of proteases of the kinin-kallikrein system, either f12 or prekallikrein (PKK) reverse the effects of C1-INH depletion with similar effects on both basal and angiotensin converting enzyme (ACE) inhibitor-induced permeability. In contrast, inhibition of coagulation factors 11 (f11) or 7 (f7) had no effect. These results suggest that the vascular defects observed in C1-INH deficiency are dependent on the kinin-kallikrein system proteases f12 and PKK, and not mediated through the coagulation pathways. In addition, our results highlight a novel therapeutic modality that can potentially be employed prophylactically to prevent attacks in HAE patients.
Introduction
The triggering mechanisms for initiation of HAE attacks are not known; however, it is generally believed that activation of the contact system and downstream effectors is a central component (Fields et al., 1983; Davis, 2005). Exposure of blood-born contact system proteases to anionic surfaces results in the conversion of the zymogen f12 to its active form f12a, which in turn activates plasma kallikrein (PKK). Activated kallikrein then cleaves high molecular weight kininogen (HK), resulting in bradykinin generation. Activated kallikrein can also act in a positive feedback loop to promote further activation of f12. In addition, f12a activates f11, which then initiates a cascade of proteolytic cleavage events resulting in thrombin generation and formation of a fibrin clot (Gailani and Renne, 2007; Schmaier and McCrae, 2007). Recent studies suggest that certain contact activation initiators, such as protein aggregates and mast cell heparin, are capable of f12 activation and the downstream components of the contact and kallikrein systems without activation of f11 or the extrinsic coagulation cascade (Maas et al., 2008; Oschatz et al., 2011). Therapeutic approaches for treating HAE have been designed to target several of these pathways, with anti-fibrinolytics, kallikrein pathway inhibitors, or replacement therapy by intravenous infusion of plasma derived C1-INH (reviewed in Cugno et al., 2009b).
Recently, significant progress has been made in the treatment of acute HAE attacks; however, these therapies have limited potential as prophylactic treatments due to a variety of factors involving pharmacokinetics, safety, and route of administration (Cugno et al., 2009b). Second generation ASOs are an emerging class of therapeutics that are highly selective and can be safely administered by weekly in-home subcutaneous injections (Henry et al., 2001). Therapeutic antisense oligonucleotides (ASOs) have demonstrated clinical activity and safety in the treatment of several diseases (Fidias et al., 2009; Bennett and Swayze, 2010; Zhang et al., 2010). Antisense therapy utilizes base pair hybridization through which ASOs selectively bind their messenger RNA (mRNA) target. The resulting DNA-RNA duplex is recognized by the ubiquitous cellular enzyme RNAseH, which mediates the catalytic degradation of the target mRNA strand and ultimately results in corresponding decreased protein levels (Zhang et al., 2010). In addition, second generation ASOs are particularly active when directed towards liver-expressed targets, such as the component of the kallikrein and coagulation systems (Zhang et al., 2010), as the liver is one of the primary organs for ASO accumulation (Koller et al., 2011).
To evaluate the contribution of the protein components of the coagulation and kallikrein pathways to vascular permeability, we generated selective ASO inhibitors of PKK, f12, f11, and f7. In addition, as 2 independently generated C1-INH knockout mice display somewhat divergent vascular phenotypes (Han et al., 2002; Oschatz et al., 2011), we also developed a potent antisense inhibitor of C1-INH to evaluate the effect of varying levels of C1-INH protein on the vasculature. Our results demonstrate that ASO-mediated inhibition of either PKK or f12 reverses both basal and captopril-induced vascular permeability, as well as vascular permeability induced by the depletion C1-INH, whereas inhibition of either f7 or f11 had no effect. These results highlight an attractive new therapeutic modality with potential as a prophylactic treatment of HAE and suggest that vascular permeability is modulated primarily via kallikrein pathway activation, whereas the extrinsic and intrinsic coagulation pathways play a minor role.
Methods
Animals
Screening for lead ASOs was performed in 8-week-old male Balb/C mice (n=4), and all other studies were performed in C57Bl/6J-Tyrc-2J (C57 albino) mice (Jackson Laboratories) according to the indicated treatment schedules (n=8). The animals were housed in micro-isolator cages on a constant 12-hour light-dark cycle. All procedures were approved by the Institutional Animal Care and Use Committee.
Oligonucleotides
ASOs were 20 nucleotides in length and chemically modified with a phosphorothioate backbone, 2′O methoxyethyl on the wings with a central deoxynucleotide gap. ASOs were synthesized using an Applied Biosystems 380B automated DNA synthesizer (Applied Biosystems) and purified as described previously (Baker et al., 1997). In order to identify the most potent PKK, f12, f11, f7, and C1-INH ASOs for animal testing, ASOs were designed and tested in primary mouse hepatocytes for their ability to suppress mRNA levels of the respective targets. Based on these results, ASOs were selected for evaluation in in vivo permeability studies in mice.
ASO dosing
PKK and coagulation factor ASOs were administered subcutaneously, twice weekly, at doses ranging from 5 to 160 mg/kg/week for 3 weeks. In combination studies with C1-INH ASO, PKK or coagulation factors ASOs were administered for 2 weeks prior to concurrent C1-INH ASO (50 mg/kg/week) administration for an additional 4 weeks. A non-specific, scrambled ASO was used as a control.
Measurement of hepatic mRNA
Mouse liver was homogenized in RLT (Qiagen) buffer containing 1% 2-mercaptoethanol. Total mRNA was prepared with the PureLink™ Total RNA Purification Kit (Invitrogen) according to the manufacturer's instructions. The amount of specific mRNA was analyzed using a StepOne™ Real-Time PCR System (Applied Biosystems). Primer and probe sequences were as follows: f7 (forward: AATGAGGAACAGTGCTCCTTTGA, reverse: TGTAAACAATCCAGAACTGCTTGGT, probe: CCCGGGAGATCTTCAAGAGCCCX); f11 (forward: ACATGACAGGCGCGATCTCT, reverse: TCTAGGTTCACGTACACATCTTTGC, probe: TTCCTTCAAGCAATGCCCTCAGCAATX); f12 (forward: CAAAGGAGGGACATGTATCAACAC, reverse: CTGGCAATGTTTCCCAGTGA, probe: CCCAATGGGCCACACTGTCTCTGCX); PKK (forward: ACAAGTGCATTTTACAGACCAGAGTAC, reverse: GGTTGTCCGCTGACTTTATGCT, probe : AAGCACAGTGCAAGCGGAACACCCX) C1-INH (forward: GAGTCCCCCAGAGCCTACAGT, reverse: TGTCATTTGTTATTGTGATGGCTACA, probe: CTGCCCTCTACCTGGCCAACAACCAX); kininogen 1 (forward: TCTGTGAGAACTTGCTCTTTTTGC, reverse: CTGGTAGGCATCATATTGACATAGATTAT, probe: CCTCTCCTGCTCACCAAGCCAATGCX); and kininogen 2 (forward: GGTAAGGGCCCCATAGTGACA, reverse: CCGATATGGGATGCACACAA, probe: AGGAGTACCACTGTGCG).
Permeability assays
Permeability assays were performed as previously described with minor modifications (Han et al., 2002). Twenty-four to forty-eight hours after the final ASO dose, male C57 albino mice were injected with Evans blue dye (30 mg/kg) in phosphate buffered saline (PBS) into the tail vein. After 30 minutes, blood was collected via cardiac puncture followed by euthanization under anesthesia; colons and both hind feet were then removed and Evans blue extracted with 1 mL of formamide overnight at 55°C and quantitated spectrophotometrically at 600 nm. In certain studies, mice were treated intraperitoneally with 20 μg of the angiotensin converting enzyme (ACE) inhibitor captopril (Sigma) to enhance basal permeability. The bradykinin 2 receptor (B2R) antagonist HOE140 (30 μg, Sigma) was administered intraperitoneally as a positive control.
Ethanol precipitation of plasma
Blood was drawn through cardiac puncture, immediately mixed with 3 volumes of ice-cold ethanol and centrifuged at 15,000 g at 4°C for 20 minutes to remove cell debris and precipitated plasma proteins. Ethanol extracts were further purified by ultrafiltration through 10-kDa molecular weight cut off filter. Ethanol-extracted Evans blue was quantified by spectrophotometry at 620 nm.
Western blotting of plasma samples
Blood was collected under anesthesia via cardiac puncture into sample tubes coated with the anticoagulant EDTA. Blood was centrifuged at 4,000 g for 15 minutes and platelet-poor plasma was collected and stored at −80°C prior to western blotting. Pooled plasma samples from all groups were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by immunoblotting with antibodies to human f12 (Accurate Chemicals), mouse PKK, human C1-INH, or mouse HK (R&D Systems).
Factor 12 and PKK chromogenic activity assay
Fifty microliters of plasma diluted 1:10 in PBS was activated with 5 μL activated partial thromboplastin time reagent (Actin® FS, Siemens) in a 96-well polypropylene microplate. After 10 minutes, 50 μL of 1 mM chromogenic substrate S-2302 (Diapharma) mixture with 500 nM of maize trypsin inhibitor for kallikrein assay, or 100 μM KALLISTOP™ (ADI) for f12 assay were added, and the amidolytic activity of the generated enzyme was determined at 405 nm. Factor 12a and kallikrein levels were measured in the linear phase of absorbance accumulation.
Human umbilical vein endothelial cell monolayer permeability
Human umbilical vein endothelial cells (HUVECs) (104 cells) we seeded onto polycarbonate inserts of a 96-well Transwell® (Corning) system (0.4 μm diameter pores) coated with 0.2% gelatin and used 5 days after plating. Each well was checked for the formation of confluent monolayer by adding Evans blue 0.2 mg/mL to the upper chamber and measuring the amount of dye passed through the insert to the lower chamber by absorbance at 600 nm. Only wells that had negligible leakage of dye through the insert (intact confluent monolayer) were taken into the subsequent experiment. Twenty microliters of plasma samples derived from mice subjected to permeability studies (described above) were added to the upper chamber and Evans blue leakage to the lower chamber was measured after 30 minutes by spectrophotometry.
Factor 12 activation assay
Plasma (5 μL) was added to 85 μL of PBS containing 1 μg/mL dextran sulfate (500 kDa) in a 96-well polypropelene microplate, and the solution was incubated for 5 minutes at room temperature. Then 10 μl of 2 mM Spectrozyme® F12a mixture with 1 mM KALLISTOP™ was added and the absorbance kinetics measured at 405 nm. Factor 12 activation was measured in the linear phase of absorbance accumulation.
Measurement of component 5a levels
Component 5a (C5a) levels in the plasma were measured by sandwich enzyme-linked immunosorbent assay. Assay plates were coated with rat anti-mouse C5a antibody (BD Biosciences) and blocked with 2% bovine serum albumin before incubation with mouse plasma samples. After washes, C5a was detected with biotinylated anti-mouse C5a antibody (BD Biosciences) followed by horseradish peroxidase conjugated streptavidin. Serial dilutions of mouse recombinant C5a protein (BD Biosciences) were used as standards.
Results
Data are presented as mean±standard deviation of the mean. The 1-tailed Student's t-test was used to compare data between groups. Statistical significance was assumed at p<0.05 (*), with p<0.01 (**) and p<0.001 (***) also noted.
Systemic administration of ASOs targeting f12 and PKK selectively reduce liver mRNA expression and plasma protein levels
In order to characterize the effects of PKK and f12 ASOs against their respective targets, ASOs were initially characterized for selectivity and potency towards various target mRNAs. Systemic administration of PKK and f12 ASOs resulted in a dose-dependent reduction (up to 90%) of PKK and f12 liver mRNA levels (Fig. 1A). Neither PKK nor f12 ASOs had any effect on the expression of non-targeted coagulation factors, thereby confirming ASO specificity (Fig. 1B). Plasma protein levels and enzymatic activity of PKK and f12 were similarly inhibited by ASO treatments (Fig 1C, D). ASOs selective for f7 and f11 also demonstrated greater than 90% reduction of target mRNA expression without any cross-reactivity with other targets (Fig. 1B). Interestingly, we observed an increase in total protein levels (zymogen and active) of f12 when PKK was inhibited, and of PKK total protein when f12 was inhibited (Fig. 1C). The increase in PKK and f12 was only observed at the protein level (not at the mRNA level in liver) and may reflect the stabilization of contact and kinin-kallikrein system components in response to decreased levels of PKK or f12, and decreased basal activation of these pathways.
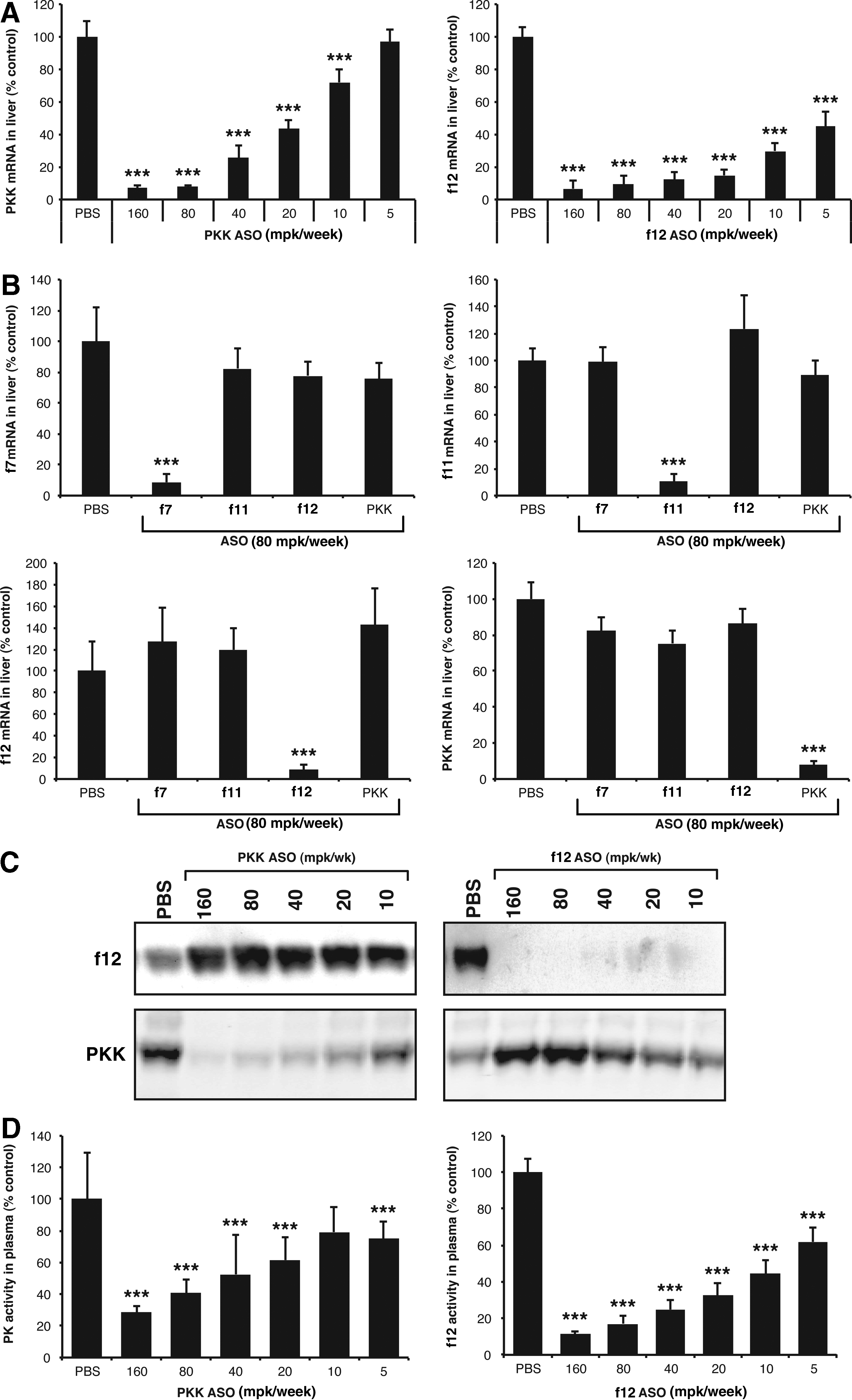
Generation of antisense oligonucleotides (ASOs) that specifically and potently inhibit prekallikrein (PKK) and coagulation factor 12 (f12).
Inhibition of PKK and f12 but not f7 or f11 resulted in decreased vascular permeability
It has been previously shown that C1-INH deficiency results in increased vascular permeability mediated via bradykinin, and that inhibition of bradykinin degradation with the angiotensin converting enzyme (ACE) inhibitor captopril further enhances permeability (Han et al., 2002). In these systems, permeability could be reversed by treatment with either human C1-INH, contact pathway inhibitors, or the B2R antagonist incatibant (HOE140) (Han et al., 2002). In order to determine the effect of inhibition of specific components of the coagulation or kallikrein pathways on vascular permeability, we evaluated both basal and captopril-induced permeability in mice treated with ASOs targeting PKK, f12, f11, and f7. After systemic treatment with ASOs, vascular permeability was determined by the quantitation of intravenously injected Evans blue dye extravasating into peripheral tissues. Parallel studies were also performed in which captopril was administered immediately prior to Evans blue injection. In both basal and captopril-induced permeability studies, the bradykinin receptor inhibitor HOE140 was included as a positive control.
ASO-mediated inhibition of PKK and f12 both significantly reduced basal vascular permeability. This inhibition was similar to what was observed with acute administration of HOE140 (Fig. 2A). Interestingly, inhibition of the coagulation factors, f7 or f11, had no effect on basal vascular permeability (Fig. 2A) demonstrating that basal vascular permeability is not significantly altered by these components of the intrinsic or extrinsic coagulation pathways.
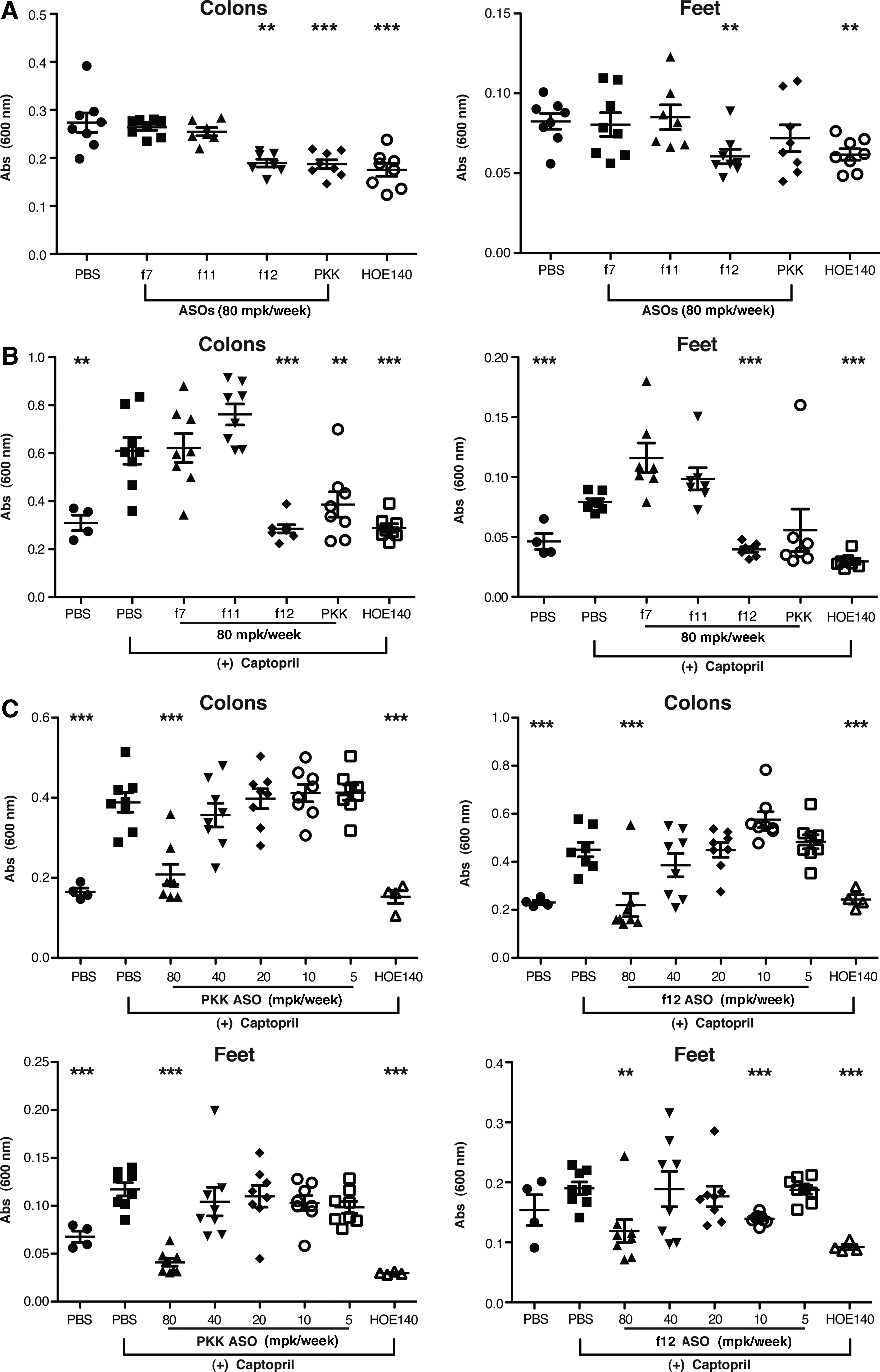
ASO-mediated inhibition of prekallikrein (PKK) and f12 results in decreased vascular permeability.
In order to explore the contribution of these pathways in a setting where vascular permeability is compromised, we performed similar studies in mice that had been treated with captopril to induce vascular leakage (Fig. 2B). Administration of captopril resulted in a significant increase in vascular permeability measured in the colon and footpad (Fig. 2B). PKK and f12 inhibition reduced vascular permeability down to basal levels in these tissues (Fig. 2B). Similar to the effect on basal vascular permeability, f7 and f11 inhibition did not affect captopril-induced vascular permeability (Fig. 2B), suggesting that the observed effects are not mediated by the coagulation system. Analysis of liver mRNA from all of the treated groups confirmed that knockdown of PKK and coagulation factors was greater than 90% (Fig. 1C). Both PKK and f12 ASOs led to a threshold-dependent reduction in captopril-induced permeability in the colon and footpad of mice (Fig. 2C).
In order to investigate the downstream effects of ASO treatment on the kallikrein pathway, we evaluated their effects on plasma HK levels and assessed kallikrein pathway activation by measuring the presence of cleaved HK in plasma. Inhibition of either PKK or f12 resulted in complete inhibition of HK cleavage as determined by immunoblot analysis of full-length HK and by quantitation of the HK cleavage fragment. A decrease in the cleavage of full-length HK levels correlated with the decrease in permeability observed with both PKK and f12 ASO treatments (Fig. 3A), suggesting that stabilization of HK contributes to the increased vascular integrity observed as a result of PKK and f12 depletion.
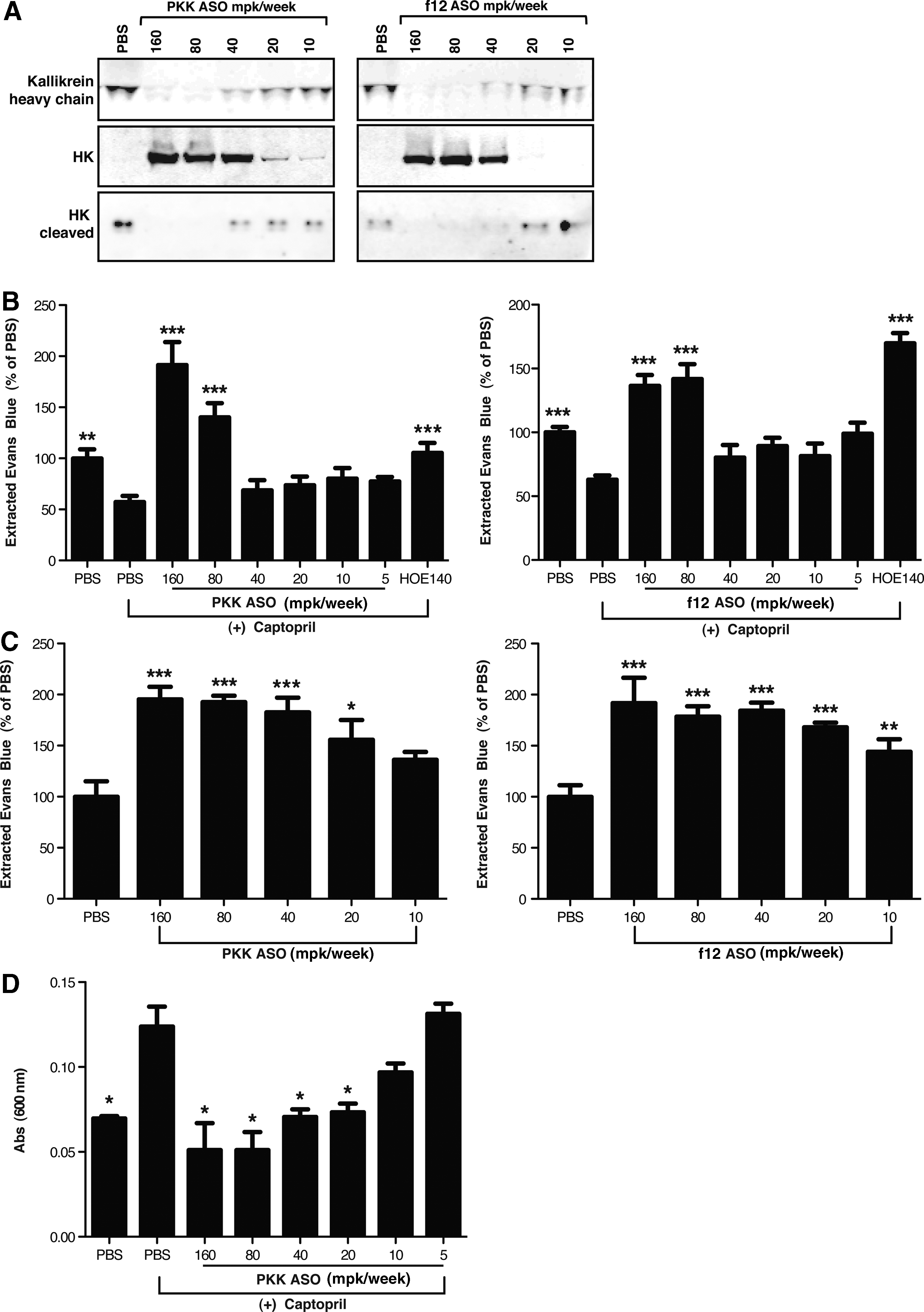
ASO-mediated inhibition of PKK and f12 stabilizes kininogen (HK) and prevents vascular leakage.
In order to expand our characterization of vascular permeability beyond individual organs, we developed a method to capture the total retention of Evans blue in the circulation using an ethanol-based extraction of free Evans blue from whole blood. In this assay, increased permeability within the vessel wall correlates well with decreased retention of Evans blue in the circulation. Addition of agents such as captopril, which promote increased leakage, result in a 40%–50% reduction in the amount of ethanol-extracted dye in the blood (Fig. 3B). A threshold-dependent increase in the amount of Evans blue retained in the blood was observed with both PKK and f12 ASO treatment (Fig. 3C), demonstrating that these factors influence the basal vascular tone in mice. Similar effects were also seen when vascular permeability was altered by prior treatment with captopril (Fig. 3B).
Previous studies have shown that plasma isolated from C1-INH deficient patients during acute attacks can induce endothelial cell leakage of a HUVEC monolayer in vitro (Bossi et al., 2009). We demonstrated that plasma derived from captopril treated mice was able to induce leakage of Evans blue through a confluent monolayer of HUVECs, and that this capacity was significantly diminished in plasma derived from captopril-induced mice treated with PKK ASO (Fig. 3D).
These data suggest that basal and captopril-induced vascular permeability is primarily mediated via the contact and kinin-kallikrein pathways and can be reversed by selective ASO-mediated inhibition of PKK and f12
C1-INH deficiency–induced increase in vascular permeability is reversed by inhibition of PKK and f12
In order to replicate the C1-INH deficiency associated with HAE, we identified a C1-INH targeted ASO that effectively reduced C1-INH mRNA expression in the liver (>95% of untreated levels) and C1-INH plasma protein levels (>95% of untreated levels) (Fig. 4A). Inhibition of C1-INH did not affect the expression of PKK, f12, or other coagulation pathway proteins measured (Fig. 4B). As observed in mice with targeted disruption of the C1-INH gene, C1-INH ASO treated mice did not exhibit spontaneous swelling events seen in HAE patients. However, C1-INH-depleted animals did show increased basal vascular permeability, demonstrated by a significant increase in Evans blue extravasation into tissues (Fig. 5A), akin to the C1-INH knockout mouse (Han et al., 2002). Increased permeability as a result of C1-INH ASO treatment was inhibited with either PKK or f12 ASO treatment (Fig. 5A). Furthermore, HK cleavage was also inhibited upon PKK or f12 inhibition (Fig. 5B). C1-INH, PKK, and f12 ASOs did not affect HK mRNA expression in liver (Fig. 5C). One possible explanation for the lack of a robust permeability phenotype in C1-INH ASO treated mice comes from our observation that as C1-INH levels are decreased by ASO treatment, there is a commensurate decrease in PKK plasma levels, potentially resulting from an increase in PKK activation and activation-dependent clearance (Fig. 5D) which may serve to counteract the effects of C1-INH deficiency.
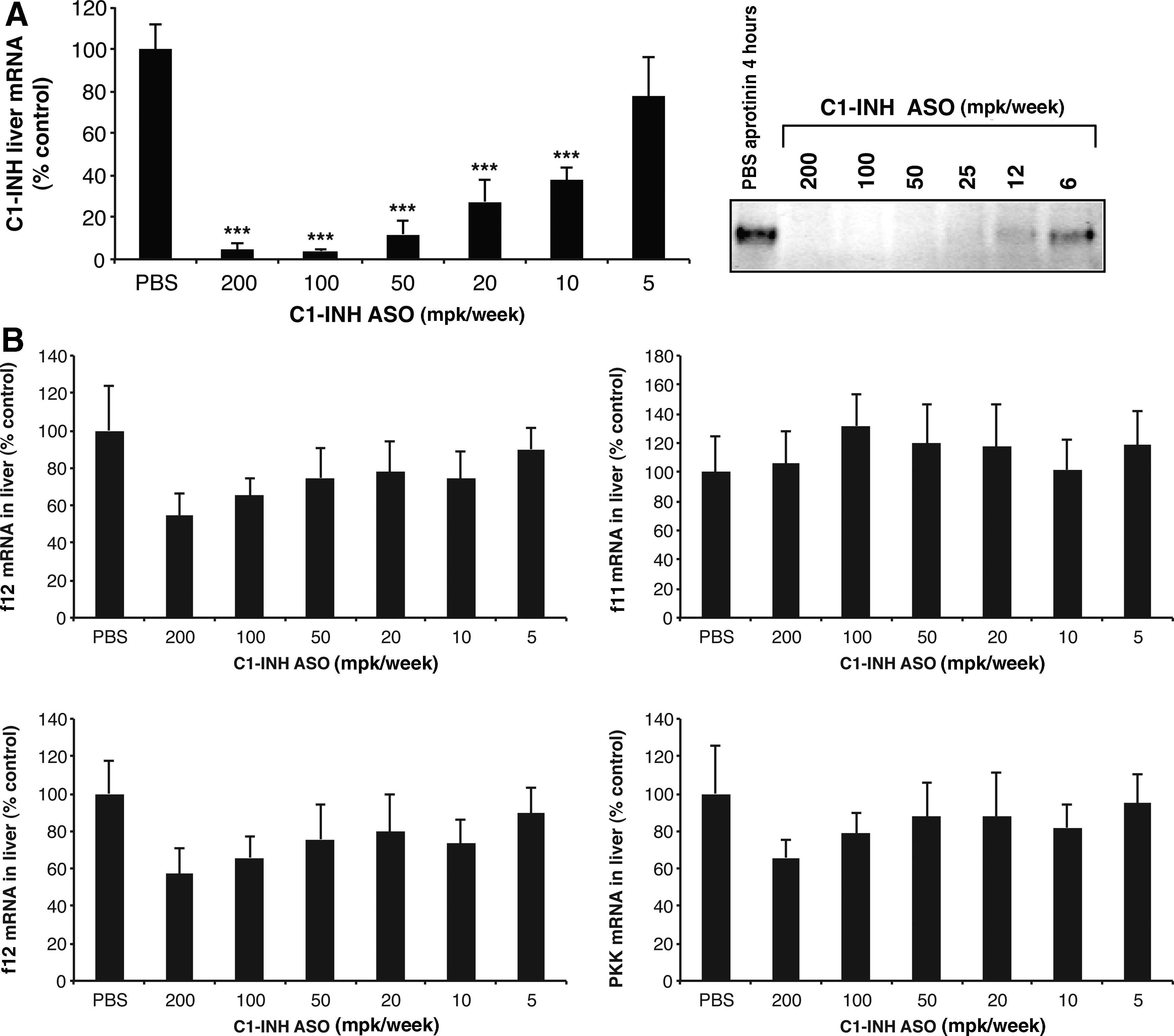
Generation of a serine protease inhibitor C1 esterase inhibitor (C1-INH)-specific ASO.
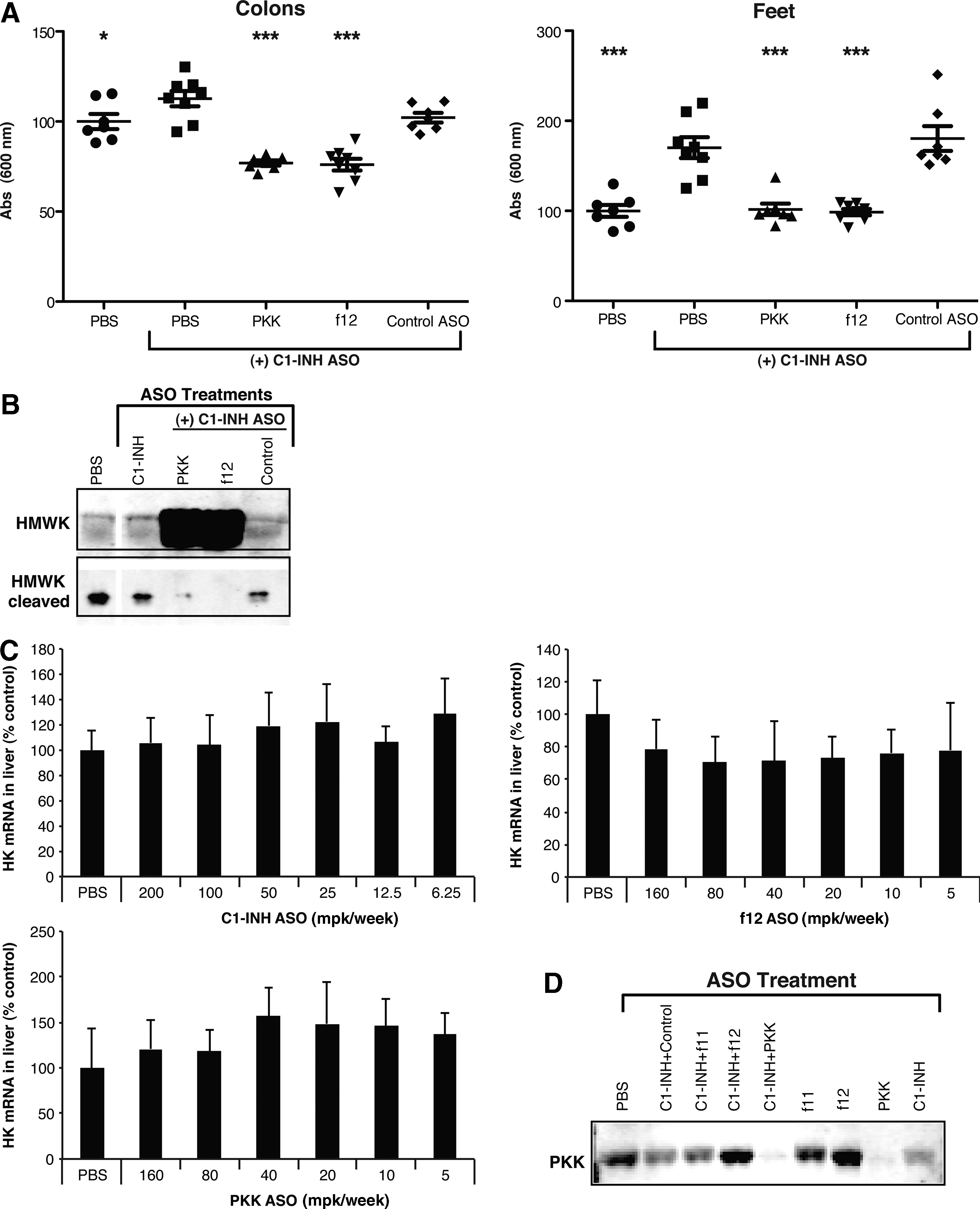
ASO-mediated inhibition of C1-INH results in increased vascular permeability and HK cleavage, both of which are reversed by PKK and f12 inhibition.
Combination of PKK and f12 ASOs synergistically reduce vascular permeability
ASOs have proven to be safe and efficacious with minimal drug–drug interactions when given in combination with other drugs for a broad range of indications (Adjei et al., 2003; Villalona-Calero et al., 2004; Geary et al., 2006). Having demonstrated the efficacy of PKK and f12 ASOs individually in reversing basal and induced rodent permeability, we tested PKK and f12 ASOs in combination to determine if there was any additive or synergistic effect in knocking down these 2 targets simultaneously. In studies performed with captopril, a synergistic reduction in vascular permeability was observed with PKK and f12 ASOs given in combination, a 50% reduction in permeability observed at doses of 20 mg/kg/week for each ASO for both colon and footpad permeability. This activity is equivalent to when 80 mg/kg/week for each ASO was given individually (Figs. 6A and 2B). Similar results were observed with the prevention of Evans blue leakage out of plasma with a significant increase in retention of dye at 20 mg/kg/week ASO treatment of PKK and f12 in captopril-induced studies (Fig. 6B). The combination of PKK and f12 inhibition had even greater effects on basal vascular permeability, where greater than 25% reduction was observed in both colon and foot permeability at concentrations as low as 5 mg/kg/week each of PKK and f12 ASOs, which was similar to that observed at 80 mg/kg week for each ASO given individually (Figs. 2C and 6C). PKK and f12 ASOs given simultaneously result in similar decreases in mRNA expression to each ASO given individually (Figs. 1A and 6D), indicating that the effects we observe are occurring at the functional level, and not due to increased inhibition of PKK or f12 expression due to the simultaneous administration of two ASOs. The synergistic effect of combined treatment of f12 and PKK ASOs is consistent with our data demonstrating that inhibition of PKK alone results in increased f12 levels, and inhibition of f12 alone results in increased PKK levels (Fig. 1C). Increased levels of PKK in f12-depleted mice of f12 in PKK-depleted mice may partially compensate for the lack of another contact factor. Upon simultaneous treatment with PKK and f12 ASOs, protein levels of both factors are reduced (Fig. 6D), which may explain more efficient contact system inhibition and downstream permeability effects (Fig 6A–C). In summary, our studies demonstrate that ASO-mediated inhibition of PKK and f12, given alone or in combination, are efficacious in reversing C1-INH deficiency and ACE inhibitor–induced vascular permeability and may serve as a potential prophylactic therapy for the treatment of HAE.

Synergistic reduction of vascular permeability with combination of PKK and f12 ASOs.
Discussion
There are currently no animal models which replicate all the symptoms of HAE. However, vascular permeability in mice has been used previously to evaluate potential HAE therapeutics and serves as an established surrogate for candidate evaluation in the preclinical setting (Han et al., 2002). Our studies demonstrate effective ASO-mediated inhibition of PKK and f12 mRNA with a concomitant decrease in protein levels and activity. Administration of PKK and f12 ASOs results in decreases in both basal and captopril-induced permeability as well as protection from vascular leakage, similar to what has previously been shown with purified plasma C1-INH protein, kallikrein inhibitors, and bradykinin receptor antagonists (Han et al., 2002). Depletion of both f12 and PKK also resulted in inhibition of f12 activation in plasma (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat) and stabilization of HK, cleavage of which results in bradykinin. This is consistent with studies indicating that the pathophysiology of HAE is due to increased bradykinin generation as a result of C1-INH dysfunction or deficiency, mediated via inappropriate activation of f12 and PKK, and not due to inhibition of the complement pathway (Fields et al., 1983; Davis, 2005) (Supplementsry Fig. S2). Initial studies involving C1-INH knockout mice focused on vascular permeability in specific organs (ear, footpad, portions of the gastrointestinal tract) and demonstrated moderate increases in vascular permeability that were further enhanced with agents such as captopril and mustard oil (Han et al., 2002). We have expanded these studies to directly quantitate Evans blue retention in circulation as a measure of the net effect of leakage into tissues. With PKK and f12 ASO treatment, alone and in combination, the retention of Evans blue dye in plasma correlates directly with protection from increased permeability observed in colon and feet.
HAE patients are not predisposed to thrombotic, coagulation, or fibrinolytic complications compared with the general population. However, it is important to note that once an angioedema attack is initiated, the coagulation, fibrinolytic, and complement cascades are all activated (Nielsen et al., 1996; Cugno et al., 1997; Cugno et al., 2009a). Elevated levels of thrombin/anti-thrombin, plasmin/alpha2-antiplasmin, and C4 activation products are all observed during the course of an attack (Nielsen et al., 1996). Interestingly, f11a–serpin complexes were not elevated (Nielsen et al., 1996), consistent with our observation that inhibition of f11 does not impact vascular permeability. Thrombin has been shown to induce vascular permeability in vivo (Lo et al., 1990) and to increase the microvascular permeability of endothelial cell monolayers (Malik and Fenton, 1992; Lum et al., 1993), an effect that correlates with reduced transendothelial electrical resistance (Tiruppathi et al., 1992). Studies using a mouse model of lung microvascular permeability demonstrated that thrombin induces permeability in this model and its effect is mediated by endothelial cell protease-activated receptor-1 (PAR-1) as PAR-1 deficiency abrogated thrombin-induced pulmonary vasoconstriction and microvessel permeability (Vogel et al., 2000). Our results with PKK and f12 ASOs and the bradykinin receptor antagonist HOE140 suggest that bradykinin generation is the major mechanism of vascular permeability in our preclinical HAE rodent models and are supported by studies showing that thrombin and bradykinin initiate discrete endothelial permeability mechanisms (Schaeffer et al., 1993).
In addition to the ACE inhibitors such as captopril, which stabilize bradykinin (reviewed in 37), studies performed with the C1-INH knockout mouse also demonstrate increased permeability that is reversed with C1-INH protein (Han et al., 2002). We attempted to replicate this phenotype using ASO-mediated inhibition of C1-INH (Fig. 4) and in doing so, we observe a similar increase in permeability that was completely reversed upon inhibition of PKK or f12 (Fig. 5), thereby demonstrating that prophylactic inhibition of PKK or f12 can reverse a C1-INH deficiency–induced phenotype in a similar manner to acute administration of recombinant C1-INH. Although inhibition of either PKK or f12 is sufficient to reverse increased vascular permeability, it is interesting that the combination of PKK and f12 resulted in synergistic protection against both basal and captopril-induced vascular leakage, suggesting the potential utility of a combination therapeutic strategy (Fig. 6).
There have been several recent advances in the treatment of HAE based on an improved understanding of the molecular mechanisms underlying the disease. These include replacement of the genetic deficiency via C1-INH administration, and targeting downstream generation or activity of bradykinin. The most successful of these have been directed at treatments of acute attacks and thus there is still a great need for an HAE prophylactic therapy. In addition to the mechanistic relevance of PKK and f12 in HAE, our rationale for targeting these proteins via antisense drugs is enhanced by the limited pathophysiology associated with deficiencies of either of these proteins in humans. Humans with complete deficiencies of PKK are asymptomatic (Girolami et al., 2010a; Girolami et al., 2010b); f12 deficiency has been correlated with a predisposition towards increased venous thrombosis (Ratnoff, 1980), though the mechanism by which this occurs remains to be elucidated, as other prothombotic factors may be involved (Girolami et al., 2004). Moreover, case-controlled studies have not found an association between f12 deficiency and adverse outcomes (Koster et al., 1994; Zeerleder et al., 1999) and severe f12 deficiencies are not associated with increased mortality (Endler et al., 2007). In addition, antisense-mediated inhibition of contact and kallikrein system proteases is not associated with bleeding risk, akin to what is observed in f12 knockout mice (Renne et al., 2005; Revenko et al., 2011). These studies, along with the safety and efficacy of the PKK inhibitor ecallantide in treating acute HAE, makes inhibition of PKK and f12 attractive targets for treatment of HAE, and provided the rationale for testing antisense inhibitors of these proteins in preclinical murine models of HAE.
Second-generation antisense drugs represent a unique opportunity for treatment of HAE. ASOs have been demonstrated to be safe and efficacious across a wide range of pathologies, including but not limited to, diabetes (Samuel et al., 2006), cardiovascular diseases (Savage et al., 2006), neurodegenerative diseases (Forte et al., 2005), and cancer (Lamoureux et al., 2011). Mipomersen, an ASO targeting apolipoprotein B for treatment of familial hypercholesterolemia (Bell et al., 2011), has recently completed four successful phase 3 trials.
The triggers for HAE attacks have long been believed to be varied and, for the most part, unpredictable. Recent work has demonstrated that mast cell heparin activates f12 in vivo resulting in increased bradykinin formation which then leads to increased vascular permeability (Oschatz et al., 2011). Edema induced in this model is driven by activation of f12 and PKK, as f12 knockout mice were resistant to heparin-induced permeability. In addition, f12 knockout mice were also protective against increased leakage as a result of challenge with the mast cell degranulator C48/80. Similar results were observed with B2R knockout mice, confirming that f12 triggered bradykinin formation, and not activation of coagulation, is the mechanism by which this process occurs (Oschatz et al., 2011). This is consistent with our results demonstrating that f12 and PKK inhibition, but not f7 or f11, provides protection against vascular leakage (Figs. 2 and 3). Furthermore, the aforementioned study used a novel strain of C1-INH knockout mice that demonstrated increased permeability of skin microvessels in response to mast cell heparin as well as exacerbation of edema in response to allergen-induced cutaneous anaphylaxis, expanding upon studies performed with the original strain of C1-INH knockout mice that showed modest increases in basal permeability that were exacerbated with edema promoting agents, such as captopril and mustard oil (Han et al., 2002).
PKK and f12 ASO-mediated treatment for HAE can potentially be targeted towards additional pathologies where the activation of prekallikrein has been shown to play a mechanistic role, such as the expansion of hyperglycemia-induced cerebral hematoma, hemorrhagic retinal and cerebral vascular permeability, and thrombosis. Studies performed in diabetic and hyperglycemic rats have demonstrated increased hematoma expansion in the presence of PKK, a highly specific process that is not replicated with bradykinin, plasmin, or tissue plasminogen activator. Local activation of PKK in the eye by carbonic anhydrase and subsequent generation of f12a has been shown to be a mechanism by which the contact system is activated, resulting in increased retinal edema in diabetic retinopathy (Gao et al., 2007).
The work we present here highlights a novel potential therapeutic approach to HAE. Our studies demonstrate that ASO-mediated inhibition of PKK or f12 provides similar protection from vascular leakage, as previously observed using C1-INH protein or bradykinin receptor antagonists. In addition to recombinant C1-INH and the B2R antagonist icatibant (HOE140), the PKK inhibitor ecallantide has recently been approved as a therapy for acute angioedema attacks, thereby confirming the utility of a PKK inhibitor in treating HAE. One of the advantages of ASO treatment via knockdown of PKK or f12 over the aforementioned compounds is that antisense drugs can be given prophylactically as a once weekly subcutaneous injection, and do not need to be administered in a hospital setting. In summary, our studies serve as preclinical proof of concept for the administration of PKK and f12 ASOs as preventative therapeutics for HAE while demonstrating a novel approach for the treatment of this disease.
Footnotes
Acknowledgements
The authors wish to acknowledge the important contributions of Sue Freier for research support and Tracy Reigle for assistance with manuscript preparation.
Author Disclosure Statement
All authors are full-time employees of Isis Pharmaceuticals. No other competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.