
Abstract
One of the hallmarks of progression of influenza virus replication is the step involving the virus uncoating that occurs in the host cytoplasm. The BM2 ion channel protein of influenza B virus is highly conserved and is essentially required during the uncoating processes of virus, thus an attractive target for designing antiviral drugs. We screened several DNA enzymes (Dzs) containing the 10-23 catalytic motif against the influenza B virus BM2 RNA. Dzs directed against the predicted single-stranded bulge regions showed sequence-specific cleavage activities. The Dz209 not only showed significant intracellular reduction of BM2 gene expression in transient-expression system but also provided considerable protection against influenza B virus challenge in MDCK cells. Our findings suggest that the Dz molecule can be used as selective and effective inhibitor of viral RNA replication, and can be explored further for development of a potent therapeutic agent against influenza B virus infection.
Introduction
The seventh segment of influenza B virus genome encodes an integral membrane protein (BM2) that acts as proton channel, expressed abundantly at the surface of infected cells and is also present in small quantities on the surface of mature virions (Sugrue and Hay, 1991). The proton channel of influenza B virus (BM2) is a unique membrane protein of 109 amino acid residues. It is essential for virus replication and oligomerizes in the viral membrane to form pH-activated proton channel and thus an attractive target for designing antiviral strategies. The current nucleic-acid-based antiviral approaches include ribozymes, DNA enzymes (Dz), short-hairpin RNA, and small interfering RNA (siRNA) which have down-regulated several target RNAs (Goila and Banerjea, 2004; Jarczak et al., 2005; Rajput et al., 2012).
DNA enzymes or DNAzymes (Dz) are synthetic catalytic deoxyribonucleic acid molecules that can be engineered to bind to their complementary sequence in the target nucleic acid through Watson–Crick base pairing and cleave at predetermined phosphodiester linkages. The catalytic efficiency is dependent on metal ions and breaks the phosphodiester bond between a purine and pyrimidine nucleotides (Santoro and Joyce, 1997; Santoro and Joyce, 1998). Several authors have shown considerable down-regulation of vital gene of many viruses including the Japanese encephalitis virus (Appaiahgari and Vrati, 2007), hepatitis B Virus (Hou et al., 2006), and HIV (Dash and Banerjea, 2004; Singh et al., 2012).
In our previous studies, we demonstrated the successful suppression of M1 gene of influenza A virus by novel DNAzymes, ribozymes and shown a significant enhancement in cleavage activity of ribozymes when used with antisense molecules (Kumar et al., 2012). The M1 gene was also significantly suppressed by using novel siRNA-chimeric-ribozyme constructs which showed synergistic catalytic efficiency and inhibited the whole virus replication in mammalian cell line (Kumar et al., 2010).
We have recently used 10-23 DNAzymes targeted to cleave at the conserved domains of the influenza A virus M2 RNA (AM2) to show successful down-regulation of M2 gene transcript in transient-expression system as well as achieved significant reduction of M2 protein. Unlike AM2, the BM2 proton conductance is completely insensitive to amantadine and rimantadine, which were the first effective drugs licensed for influenza treatment. Thus, in the present study, we sought to use the novel DNAzymes with the hope that it will facilitate the target specific cleavage of BM2 gene transcript of influenza B viruses. To the best of our knowledge, this is the first report of the application of a DNAzyme for efficiently down-regulating the BM2 gene translation and subsequently inhibiting the replication of influenza B viruses.
Materials and Methods
Cells and viruses
MDCK cells were purchased from National Centre for Cell Sciences, Pune, India and maintained in minimum essential medium (Sigma), supplemented with 10% fetal calf serum (FCS) and antibiotics (100U/mL of penicillin and 100 μg/mL of streptomycin). The influenza B virus strain (B/Yamagata/1/73) used for this study was propagated in MDCK cell line at 37°C/5% CO2 for 72 hours in our laboratory as per standardized protocol. All the stocks were titrated by hemagglutination assay (HA) test. Briefly, the HA test was carried out in U-bottom 96 well plates. Serial two fold dilutions of the virus sample was mixed with equal volume of 0.5% suspension (vol/ vol) of chicken RBC and incubated at 4°C for 30 minutes. The wells containing adherent, homogenous layer of RBCs were considered as positive. The culture supernatants, after obtaining the HA titer were collected and stored at −80°C for further use. All experiments with virus were done in a basic safety level-2 cabinet.
Plasmid construct and in vitro transcription of BM2 gene
The viral RNA was isolated, total cDNA prepared using ImProm reverse transcription kit (Promega), and the 330 nucleotides BM2 gene (nucleotide residues 771–1,100) was amplified using gene specific primers (Odagiri et al., 1999) having restriction sites for KpnI and XhoI in the forward and reverse primers respectively. The BM2 gene was cloned in mammalian expression vector pSecTag2B and plasmids were sequenced to check the authenticity of the recombinant clone (BM2-pSecTag2B). The clones were linearized with suitable restriction enzymes and subjected to in vitro transcription using T7 RiboMAX Express Large-Scale RNA Production System (Promega). Non-structural gene 1 (NS1) of influenza B virus was also cloned in pcDNA3.1 (+) vector for assessing the efficacy and specificity of Dz and named NS1-pcDNA3.1 (+).
Selection of cleavage sites and designing of DNAzymes
Secondary structures of BM2 gene were obtained and analyzed using the M-fold RNA folding program for nucleic acid folding and hybridization prediction (www.bioinfo.rpi/applications/mfold). The conserved looped regions having the cleavage sites (phosphodiester bond between a purine and pyrimidine, 5′-AU-3′) were selected and four DNAzymes (Dzs) having 10-23 catalytic motifs were designed against these loops at various regions in BM2 RNA (Fig. 1a). The DNAzymes, Dz104, Dz146, Dz209 and Dz257, named based on their respective cleavage position (Table 1), were chemically synthesized and obtained from Sigma Genosys. The conserved 15-nt (5′- GGCTAGCTACAACGA-3′) 10–23 catalytic motif was flanked on both sides by substrate binding arms of the Dz that were made complementary to the target RNA. A mutant Dz possessing a single nucleotide substitution (G to C) in the conserved catalytic motif was also synthesized (Goila and Banerjea, 2001). The controls having scrambled arms were also synthesized to demonstrate the target sequence specificity.
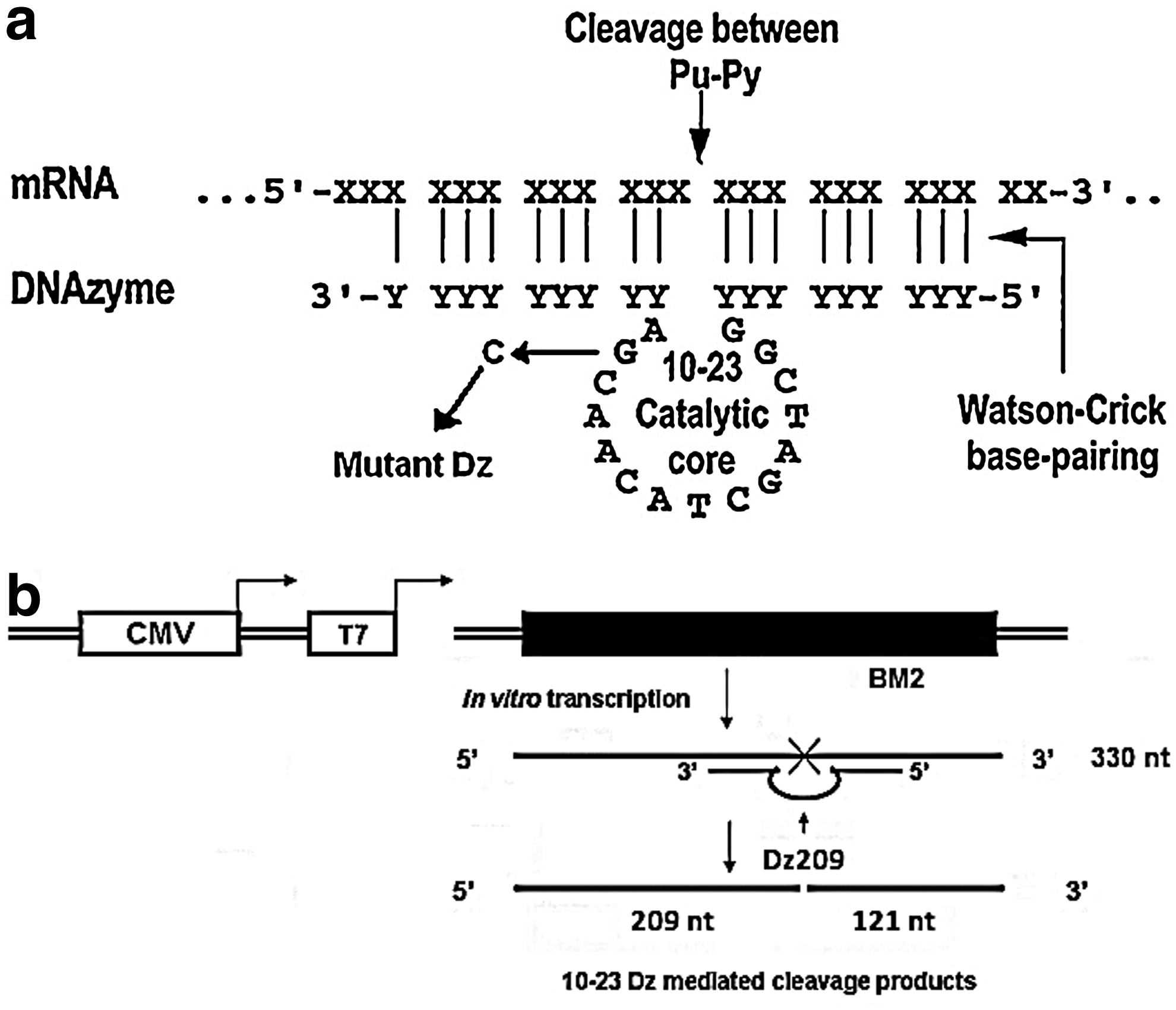
The 10-23 DNA enzymes (DNAzymes, or Dzs) were designed using the secondary structures of BM2 RNA.
The mutated residue in the mutant Mt-Dz209 is underscored.
In vitro cleavage of the substrate RNA by DNAzyme
The DNAzyme and the in vitro synthesized RNA were heated separately for 2 minutes at 95°C followed by snap-chilling on ice. The cleavage reaction was carried out by using equimolar amounts (100 pmol each) of substrate RNA and Dz in a final volume of 10 μL in a buffer containing 50 mM Tris–HCl, pH 7.5 in the presence of 10 mM MgCl2 at standard conditions (Shahi et al., 2001) for 2 hours at 37°C (Fig. 1b). The BM2 transcripts were further subjected to cleavage reactions in the presence of 10–50 mM concentrations of MgCl2.
RT-PCR and real-time RT-PCR
Reverse-transcription polymerase chain reaction (RT-PCR) was performed for full-length amplification of BM2 gene to analyze the cleavage by specific DNAzymes. The specific cleavage was also analyzed using TaqMan probes (Invitrogen) synthesized to hybridize to the Dz209 cleavage region in the substrate RNA. The probes were designed based on the genome sequence and structure of influenza B virus (B/Yamagata/1/73) messenger RNAs using RNAdraw, Primer Premier 5.0, and the National Center for Biotechnology Information Basic Local Alignment Search Tool.
The real-time RT-PCR consisted of One-step RT-PCR 2X-Super Mix (Ambion), 0.1 μM of each forward (Fd: AATCCAAATAAAGAGACAATAAAT) and reverse (Rv: GGTCACTCAATCTCTCCATGTTGT) primers and probes (FAM′: AGTACTTCCT TTATTGTTTCTTTG-BHQ1), 5 μL of template and RNAse-free water upto a volume of 25 μL. The reaction was carried out with the following cycling conditions: 50°C for 30 minutes for RT step, 95°C for 5 minutes followed by 40 cycles at 95°C for 15 seconds and 55°C for 30 seconds. All the experiments were performed in triplicates and the data was expressed as means±standard deviation (SD).
Cotransfection of DNAzymes and plasmids
MDCK cells were maintained in 6 well plates (0.5×106 cells/well) in DMEM media containing 10% heat inactivated FCS, 2mM
We assessed the efficacy and specificity of Dz209 by analyzing the effect of irrelevant Dz against NS1 gene (NS1-Dz) or mutant Dz (Mt-Dz209) on BM2 gene expression and effect of Dz209 on the gene expression of NS1 gene respectively. MDCK cells treated with 100 pmol Dz209 or Mt-Dz209 or irrelevant Dz against NS1 (NS1-Dz) were transfected with BM2-pSecTag2B clone. For specificity analysis, NS1-pcDNA3.1 (+) clone with or without 100 pm Dz209 was transfected in MDCKcells. Total cellular RNA was isolated, after 24 hours of transfection, as previously described and subjected to conventional PCR and real-time RT-PCR analysis (Kumar et al., 2012). The glyceraldehyde-3-phosphate dehydrogenase was amplified as an internal gene control for each sample. The experiment was performed in triplicates and the data was expressed as means±SD.
DNAzyme transfection and virus challenge
MDCK cells, at 70% confluency, were transfected with 1, 3, and 5 μg of Dz using Lipofectamine 2000 as described earlier. Influenza B virus strain [B/Yamagata/1/73] was inoculated as described earlier at a multiplicity of infection (MOI) of 0.1 at 8 hours posttransfection. Briefly, the transfected cells were washed twice with plain media and 100 μL of the virus in infection medium (DMEM containing 0.3% bovine serum albumin, 10mM Hepes, 100 units/mL penicillin, and 100μg/mL streptomycin) was added to them. The cells were incubated with the virus for 1 hour at 37°C and then 2 mL infection medium containing 2μg/mL of TPCK-treated trypsin was added to each well of the 6-well plate. The cells were then incubated at 37°C under 5% CO2 environment. The supernatant of the infected culture was harvested at 24 hours post infection for the determination of virus titer and the cells were lysed using Ribozol reagent (Amresco) for RNA extraction. The extracted RNA was quantified by spectrophotometric analysis at 260 nm and real-time RT-PCR was performed as described earlier.
The cycle times (Ct value) were analyzed and the Ct value that varied by>1 unit between the triplicates were discarded. The average of the triplicate Ct values was calculated and normalized to the Ct value of β-actin (Sohail et al., 2003) and expressed as means±SD. The cytopathic effect (CPE) of the viruses on the cells transfected with Dz209 was observed at 24 hours post infection and effect of the Dz on virus multiplication was analyzed by subjecting the cell supernatant to hemagglutination assay as describer earlier. Ten-fold dilutions of the culture supernatants were prepared and used for plaque assay as described earlier (Huprikar and Rabinowitz, 1980). The results were expressed as mean plaque-forming unit per mL±SD.
Sodium dodecyl sulfate polyacrilamide gel electrophoresis and immunoblotting
At 24 hours post infection, the cells were lysed using mammalian cell lysis buffer [0.1M NaCl, 0.01M Tris Cl (pH 7.6), 0.001M EDTA (pH 8.0), 1mM protease inhibitor cocktail (Amresco), 100 μg/mL phenylmethylsulfonyl fluoride]. The protein concentration was determined by Bradford assay (Amresco) and whole cell lysate was fractionated on 12% polyacrylamide for western blotting. The blot was developed using antibody against BM2 protein of influenza B virus raised as per method described by Odagiri et al. (Odagiri et al., 1999) using enhanced chemiluminescence (ECL) reagent as per manufacturer's instructions.
Statistical analysis
The data were collected from at least 3 independent experiments. All statistical analyses were performed using Prism software (Version 5). Descriptive statistics were expressed as mean±SD. Statistical significance was determined by using Student's t-test and one-way ANOVA analysis. The p values less than 0.05 were considered statistically significant.
Results
Construction of DNA enzymes and selection of target sites in the BM2 gene
Several predicted secondary structures of the entire RNA of BM2 gene was obtained through the M-fold software. Four DNAzymes (Table 1) with the lowest free energy (ΔG) values against the nucleotides position at 104 (Dz104), 146 (Dz146), 209 (Dz209), and 257 (Dz257), all designed to cleave an unpaired purine and paired pyrimidine residue (AU) in the presence of a divalent cations, were designed and synthesized commercially. Of the four DNAzymes, Dz209 gave considerable down-regulation of the BM2 gene in vitro (Fig. 2) while Dz104, Dz146, and Dz257 showed insignificant inhibition (data not shown). The mutant version of the Dz209 (Mt-Dz209), synthesized by a single base substitution (G to C) in the conserved catalytic motif, and controls having scrambled binding arms were evaluated to demonstrate the target sequence specificity of DNAzyme.
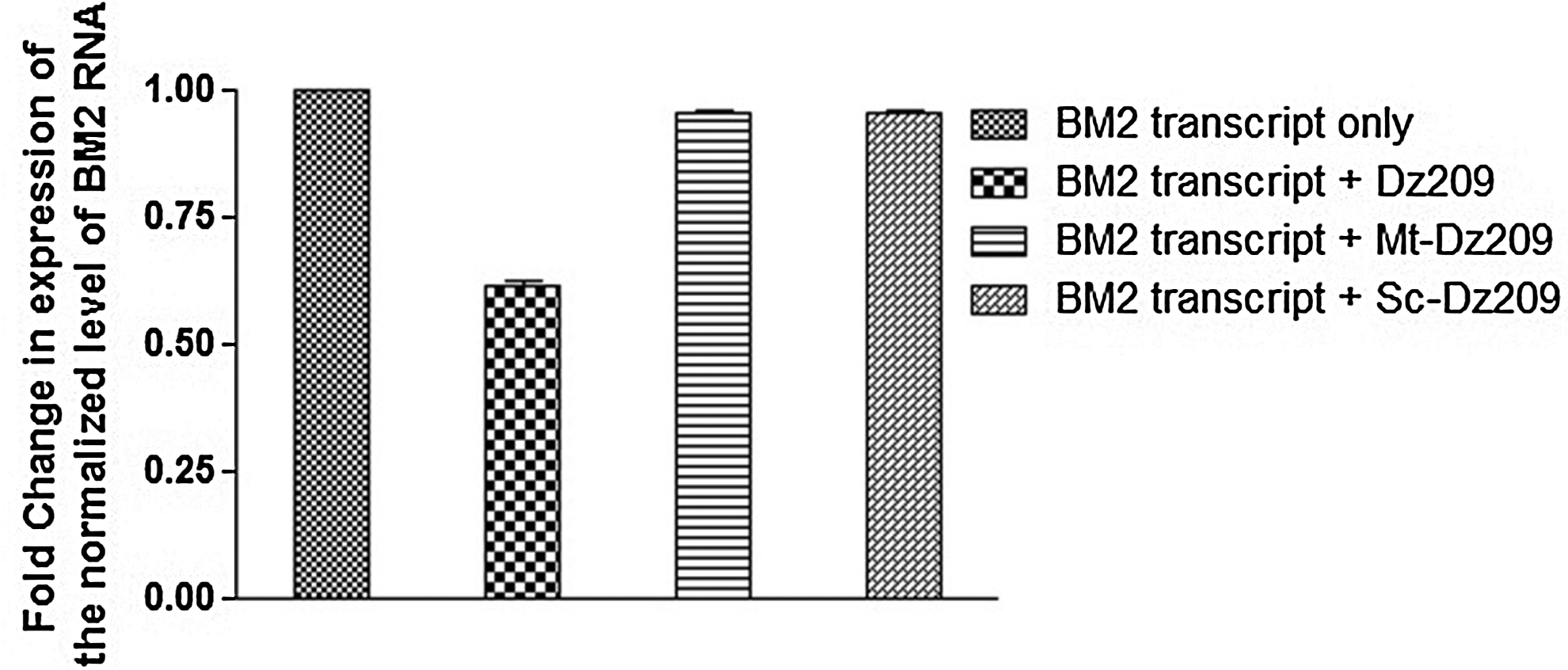
The BM2 gene was in vitro transcribed using the T7 RiboMAX TM Express Large-Scale RNA Production System (Promega). The full-length BM2 transcripts were subjected to cleavage with Dz209. The real-time reverse-transcription polymerase chain reaction (RT-PCR) based on the TaqMan chemistry was performed using primer and probes targeted to bind to the Dz209 cleavage site on BM2 RNA and detect the fold change in expression of BM2 gene in Dz209-treated samples. The experiments were performed in triplicate and the error bars indicate standard deviation (SD).
Sequence-specific in vitro cleavage of BM2 RNA by Dz209
The BM2-pSecTag2B clones were linearized and subjected to in vitro transcription using the T7 RiboMAX Express Large-Scale RNA Production System (Promega). The 330 nucleotide long BM2 RNA was specifically cleaved in the presence of Dz209 (100 pmol each) under standard conditions as described by us earlier. Two cleaved products of the expected size (209 nt and 121 nt) were also obtained which was validated by the real-time RT-PCR assay designed for the Dz209 cleavage region (Fig. 2). The disabled DNAzyme carrying the single point mutation in its catalytic motif and the Dz with scrambled arms failed to cleave the target RNA. The catalytic activity of the Dz209 was found to be weakly dependent on the magnesium ion concentration (10–50mM) when carried out under in vitro conditions and showed catalytic efficiency from 39% at 10 mM MgCl2 to 45% at 50 mM MgCl2 (Fig. 3).
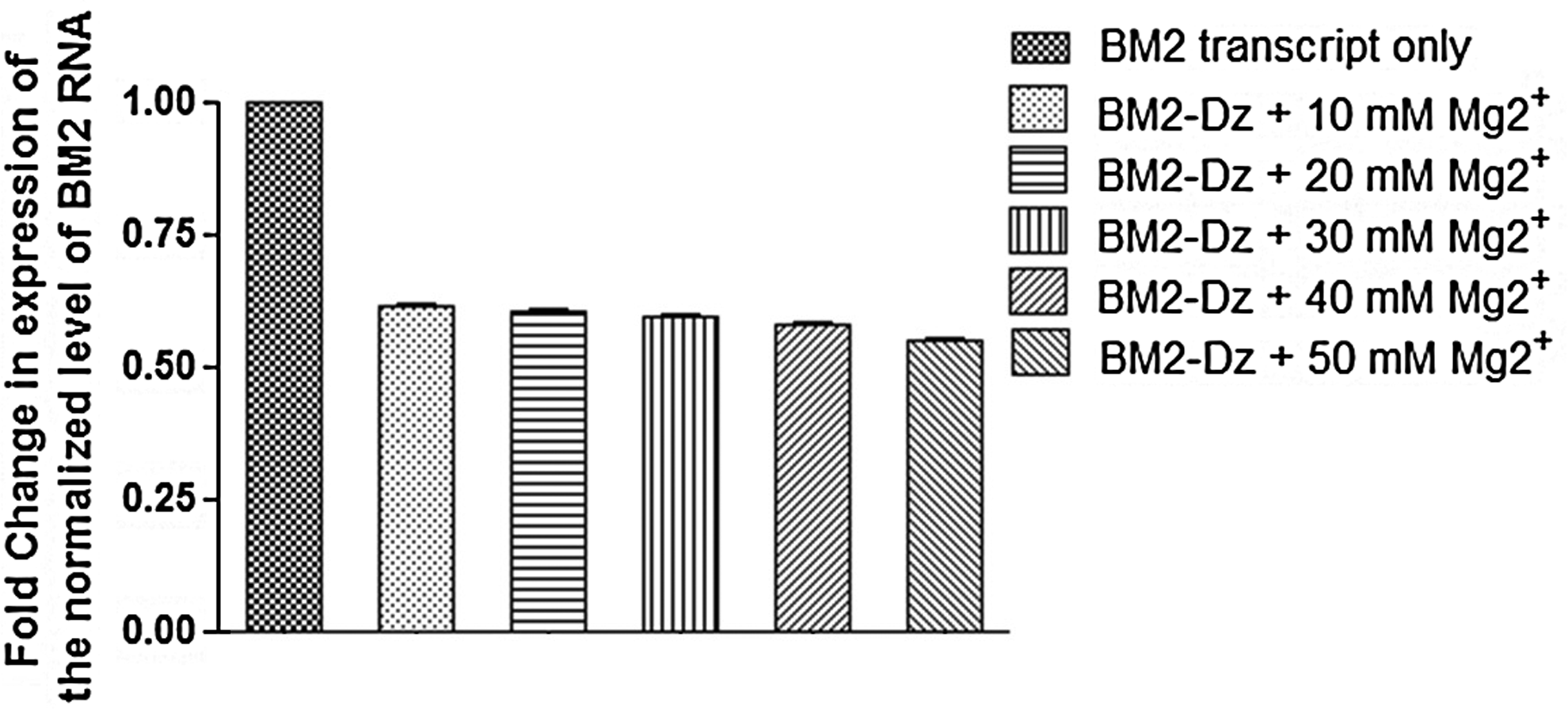
The BM2 gene was in vitro transcribed using the T7 RiboMAX TM Express Large-Scale RNA Production System (Promega). The full-length BM2 transcripts were subjected to cleavage with Dz209 in the presence of increasing concentration (10–50 mM) of MgCl2. The real-time RT-PCR was performed to detect the fold change in expression of BM2 gene in Dz209-treated samples. The experiments were performed in triplicate and the error bars indicate SD.
Down regulation of BM2 gene expression in MDCK cells
The BM2-pSecTag2B clone was commercially sequenced for confirmation of the BM2 coding sequence in correct orientation. BM2 clone and DNAzyme cotransfected MDCK cells were harvested 24 hours posttransfection for analysis of intracellular levels of BM2 gene. Out of the set of 4 Dzs, Dz209 was found to be effective against the target gene. Approximately 45% decrease in RNA levels of BM2 gene was observed post Dz209 treatment by RT-PCR (Fig. 4a) and real-time RT-PCR analyses (Fig. 4b). The result was found to be almost constant at 100 pmol and higher concentration of Dz209.

RT-PCR analysis shows intracellular reduction of BM2 RNA in Madin Darby canine kidney (MDCK) cells treated with Dz209.
The inability of the mutated Dz (Mt-Dz209) or irrelevant Dz (NS1-Dz) against NS1 gene or Dz with scrambled binding arms, to down regulate the expression of BM2 gene confirmed the efficacy of Dz209 (Fig. 4c). To further validate the specificity of Dz209 designed by us, we analyzed the effect of Dz209 on the gene expression of NS1 gene, used as unrelated viral RNA control. The level of NS1 RNA in the 100-pm Dz209–treated and NS1-pcDNA3.1 (+)–transfected MDCK cells was almost similar to the RNA levels in the MDCK cells transfected with the NS1-pcDNA3.1 (+) clone alone (Fig. 4d). This showed that the Dz was specific for the BM2 gene and efficiently inhibited the target gene expression. The experiments were performed in triplicate and the error bars in the figures indicate standard deviation.
Variable protection against virus challenge with Dz209 in MDCK cell line
The cleavage potential of the Dz209 was further validated by transfection studies in MDCK cells by performing real time RT-PCR analysis and SDS-PAGE followed by western blot analysis. The cells, after 8 hours of transfection, were given virus challenge at MOI of 0.1. The real-time PCR assay, based on TaqMan chemistry, was performed for assessing the extent of inhibition in virus replication. The ΔΔCt values were calculated and compared with the RNA level of BM2 in untreated virus–infected cells. The PCR analysis showed a significant 52% down-regulation in cells treated with 5 μg of Dz209 (Fig. 5a). The Mt-Dz209, however, failed to provide any protection against the virus challenge thereby proving the specificity of the designed DNAzyme against target RNA. The Dz209 with scrambled arms showed no down regulation revealing specificity of the DNAzyme towards the target RNA.

Different concentrations of Dz209 were given to MDCK cells against the virus challenge. Twenty-four hours post virus challenge, the cells were harvested for RNA isolation and cell lysate preparation.
The intensity of bands corresponding to viral proteins also revealed that the DNAzyme considerably down-regulated the BM2 protein expression (Fig. 5b) and thus inhibited the influenza virus replication in mammalian cell line. A significant reduction in viral protein was observed when subjected to 5 μg of Dz209. A considerable reduction in cytopathic effect (CPE) was observed 24 hours post infection thus showing inhibition of whole virus replication in MDCK cells (Fig. 6). The plaque assay also revealed substantial inhibition of viral replication in cells transfected with Dz209 (Fig. 7). All the experiments were performed in triplicate and the error bars in Fig. 5a and Fig. 7 indicate SD.
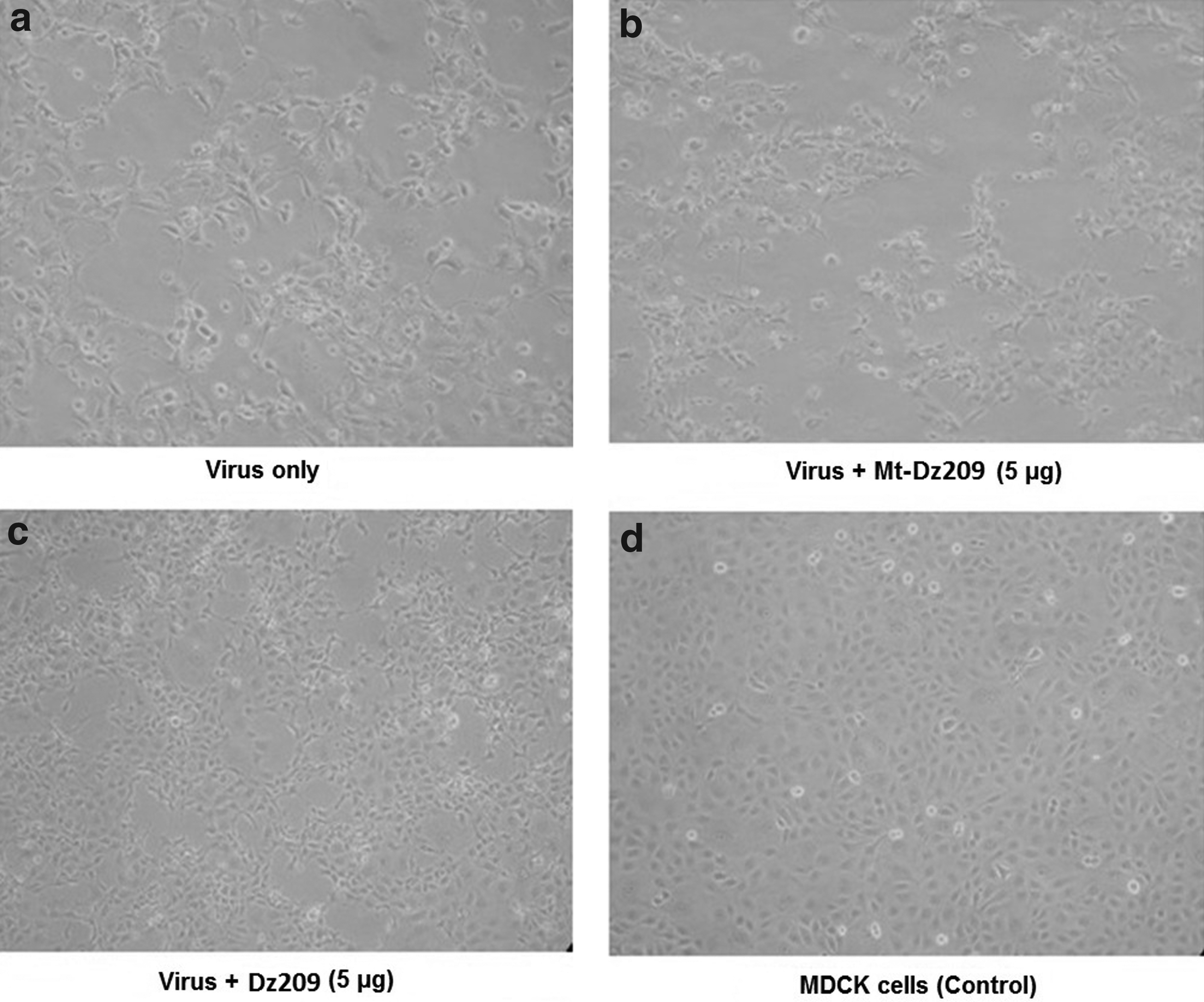
Treatment of MDCK cell line with wild-type DNAzyme (Dz209) inhibited the multiplication of virus (infected at a multiplicity of infection of 0.1) and reduced its cytopathic effect (CPE). The figures show the CPE at 24 hours post infection.
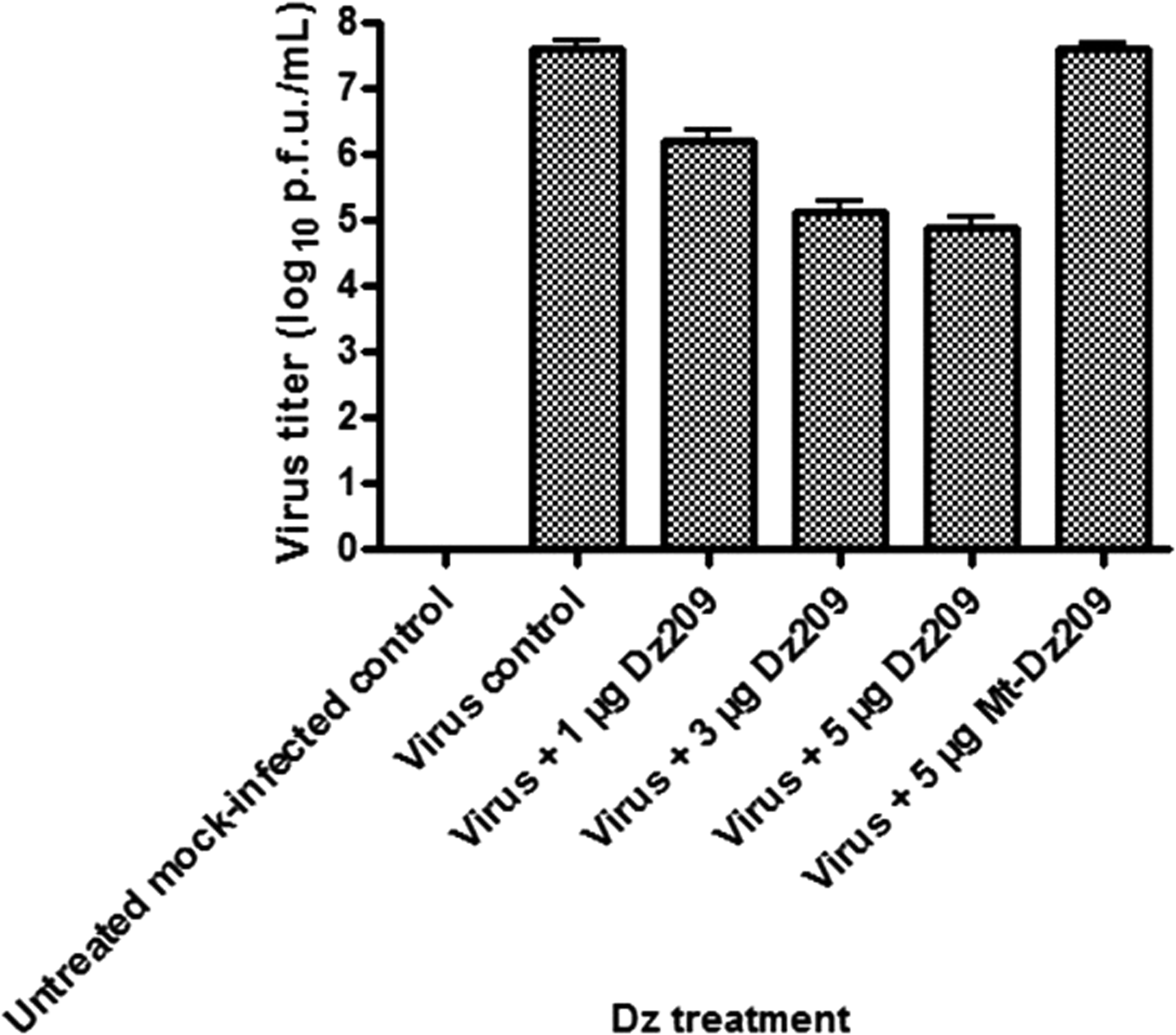
The cells were collected 24 hours post virus challenge, and virus titration was performed by plaque assay. The virus titer in the cells treated with different concentrations of Dz209 was significantly reduced in comparison with virus infected control or mutant Dz groups. The experiment was performed in triplicate and the error bars indicate SD.
Discussion
The matrix protein (BM2) of influenza B virus is known for its ion-channel activity that serves an important function in the favor of the viruses for their replication and propagation in host cells. The need for an annual influenza vaccine formulation, owing to the continuous mutation in its genome, calls for an effective strategy that can restrain the virus replication in host cells. Previous reports from our laboratory have demonstrated the use of 10-23 DNAzymes targeted against the M1 gene of influenza A viruses, which plays crucial role in viral life cycle (Kumar et al., 2012). The M1 gene was effectively down regulated by our novel DNAzymes which gave a significant protection against the A/PR/8/34-H1N1 strain of influenza A virus. We achieved excellent results with both fulllength (788 nt) and truncated (356 nt) M1 RNA.
In the present study we have targeted the BM2 gene transcript of influenza B virus by specific 10-23 DNAzymes to inhibit the propagation of virus in the host cells. Several DNAzymes with the lowest ΔG values were designed to cleave the BM2 transcript of influenza A viruses. Only one out of 4 DNAzymes (Dz209) showed significant down regulation while others failed to give satisfactory results. The result with whole virus challenge was better (52% down-regulation) as compared to the in vitro results (45% down-regulation). This could be attributed to the fact that the DNAzymes are designed based on software predicted RNA structures and during in vitro or ex vivo experiments all cleavage sites may not be accessible for efficient cleavage by these molecules. Authors have demonstrated that DNAzymes with longer arms are more efficient in cleavage activity as compared to shorter arms (Oketani et al., 1999), however we achieved equivalent results with shorter arms in our study. The down-regulation of the BM2 gene was found to be effective in a dose-dependent manner and was best at 50 mM MgCl2 concentration, which is comparable to other published reports on other viruses (Santoro and Joyce, 1998; Sood et al., 2007). The study was conducted with both wild type and mutant Dz to elucidate that the BM2 gene suppression was highly specific and that the viral gene expression could be modulated accordingly. The failure of mutant Dz209 and Dz with scrambled binding arms to down-regulate BM2 gene transcript further validated our findings.
We have reported concordant results with other DNAzyme studies as potent antiviral agents (Dash and Banerjea, 2004; Takahashi et al., 2004). Our study revealed that the designed Dz209 could not only suppress the BM2 transcript but also conferred significant protection against the virus challenge by the B/Yamagata/1/73 strain. The inhibition of influenza B virus replication was revealed by the marked reduction in viral CPE and plaques. We further analyzed the effect of Dz209 against NS1 gene and found no significant difference in the levels of NS1 RNA levels, thereby suggesting the highly specific nature of our designed DNAzyme.
To the best of our knowledge, no such study has been done on BM2 gene of influenza B viruses. Although the percent down-regulation was less as compared to its counterpart in influenza A virus (data not shown), yet the study has revealed new strategies for effective management of influenza B infections and has paved new path for effective usage of these excellent posttranscriptional gene silencing tools for therapy against influenza infections.
We conclude that the highly mutating virus such as influenza can be managed efficiently if their conserved genes are targeted. The novel DNAzymes, targeted against influenza B viruses, used in this study have successfully shown the down-regulation of one of the vital genes (BM2) responsible for virus replication. Our results provide a validation that the BM2 gene transcript of influenza B viruses could be an effective target using 10-23 DNAzymes to develop novel therapies against influenza infections.
Footnotes
Acknowledgments
We thank the Indian Council of Medical Research, government of India, for providing financial support [F. No. 80/666/ECD-I, 2010] to the author for successful completion of the study. We also acknowledge our laboratory staff Vishwamohan, Mahesh Kumar, and Satish for providing the technical support during the study.
Author Disclosure Statement
No competing financial interests exist.