
Abstract
Posttranslational modifications on proteins can serve as useful biomarkers for disease. However, their discovery and detection in biological fluids is challenging. Aptamers are oligonucleotide ligands that demonstrate high affinity toward their target proteins and can discriminate closely related proteins with superb specificity. Previously, we generated a cyclophilin B aptamer (M9-5) that could discriminate sera from pancreatic cancer patients and healthy volunteers with high specificity and sensitivity. In our present work we further characterize the aptamer and the target protein, cyclophilin B, and demonstrate that the aptamer could discriminate between cyclophilin B expressed in human cells versus bacteria. Using mass-spectrometric analysis, we discovered post-translational modifications on cyclophilin B that might be responsible for the M9-5 selectivity. The ability to distinguish between forms of the same protein with differing post-translational modifications is an important advantage of aptamers as tools for identification and detection of biomarkers.
Introduction
P
PTMs on individual proteins are typically identified using mass-spectrometric techniques and validated using conventional biochemical methods such as western blots, site-directed mutagenesis, and activity assays. The identification of PTMs in biological samples, which are complex mixtures of proteins, becomes more challenging. The protein mixtures need to be resolved by their charge and size [2-dimensional (2-D) gel electrophoresis] before they are subjected to the mass-spectrometric (MS) analysis. This often results in the loss of signals from proteins that are relatively rare and consequently are missed during the mass-spectrometric detection. Multidimensional protein identification technology (MudPIT) is an improved method that combines non-gel, 2-D liquid chromatographic separation of the protein components with mass-spectrometric identification and has been used for the discovery of PTMs in biological specimens (McDonald and Yates, 2002). Additional techniques, like surface-enhanced Raman spectroscopy are also available for PTM discovery in biological samples (Sundararajan et al., 2006). However, there is a gap between the discovery of PTMs and the ability to detect them in biological samples due to the lack of detection reagents, such as antibodies, with suitable affinity and specificity for these protein-PTMs (BLOW, 2007; Kazanecki et al., 2007; Taussig et al., 2007; UHLEN, 2007).
Aptamers demonstrate specificity and affinity that can parallel good monoclonal antibodies and, as detection reagents, are an attractive alternative to antibodies. Aptamers are short DNA or RNA molecules that are selected to bind their target molecules by an in vitro selection process called SELEX (systematic evolution of ligands by exponential enrichment) (Ellington and Szostak, 1990; Tuerk and Gold, 1990). Aptamers demonstrate remarkably high target affinity with typical equilibrium dissociation constants (Kd) in the pico to nanomolar range and outstanding specificity that can distinguish even between closely related molecules with small structural differences. For example aptamers have been selected that can discriminate between targets with subtle differences, such as isozymes (Conrad et al., 1994), proteins with point mutations (Ishizaki et al., 1996; Held et al., 2007), small molecules that differ only by a methyl group (Jenison et al., 1994), minor protein conformational changes (Zichel et al., 2012), and proteins with or without specific acetylation patterns (Williams et al., 2009). Unlike antibodies, aptamer generation does not require a biological system and is not constrained by the need to generate an immune response. Additionally, because SELEX is an in vitro process, steps like counter selection can be easily incorporated in the selection scheme to generate aptamers tailored to distinguish between closely related targets, such as proteins with different PTMs. These virtues of aptamers, along with the relative ease with which they can be chemically synthesized, make them ideal to probe for structural differences in proteins that might exist between normal and diseased states. SELEX can be performed against complex mixtures of proteins (even whole cells) for the purpose of identifying aptamers that distinguish between those mixtures. Unlike the traditional biomarker discovery approach that involves target identification by mass-spectrometric methods followed by generation of detection reagents, the aptamer approach does not rely upon the precise identification of the target since the aptamers generated can be used themselves as detection reagents. Thus, aptamers can be used for the dual purpose of biomarker discovery and detection.
We have recently used this approach to find biomarkers for pancreatic cancer (Ray et al., 2012). An in vitro positive/negative SELEX strategy was devised to identify RNA aptamers that can detect structural differences between the secretomes of pancreatic cancer and noncancerous pancreatic epithelial cells. We identified an RNA aptamer (M9-5) that differentially bound the cancerous and non-cancerous secretomes. This aptamer further discriminated between the sera of pancreatic cancer patients and healthy volunteers with high sensitivity and specificity. We utilized biochemical purification methods and mass-spectrometric analysis to identify the M9-5 target as cyclophilin B (CypB). CypB could be detected in the sera using enzyme-linked immunosorbent assay (ELISA), and levels were elevated in pancreatic cancer patients compared to the healthy volunteers. The correlation between M9-5 binding and CypB levels by ELISA values had high statistical significance but was imperfect (Ray et al., 2012). This led us to question whether M9-5 recognizes different epitope(s) on CypB than those recognized by the antibody.
To address this question, we first present unequivocal evidence that the target for M9-5 is CypB by demonstrating direct binding of M9-5 to purified human CypB in vitro. Interestingly, we found that M9-5 could discriminate between human CypB that has been expressed in mammalian cells versus bacteria. Proteins expressed in mammalian cells are often modified by PTMs that are either absent or different in the bacterial system. This directed us to investigate the PTMs present on CypB that might be responsible for the aptamer specificity.
Materials and Methods
Cylcophilin B proteins
CypB is secreted by mammalian cells and contains a signal sequence (Met 1–Ala 33) in the N-terminus. For expressing the recombinant protein in human embryonic kidney (HEK293) cells, the open reading frame encoding the mature form of human CypB (Asp 34–Ala 212) was fused with a signal peptide at the N-terminus and a polyhistidine tag at the C-terminus. The C-terminal endoplasmic reticulum (ER) retention motif (Ile 213–Glu 216) was deleted, and the secreted protein was purified from the conditioned media. The proteins (samples 1 and 2) were purchased from Creative Biomart (PPIB-652H) and Sino Biological Inc. (11004-H08H) (sample 3). Human CypB (Leu 26–Glu 216) expressed in the bacterial Escherichia coli system (sample 5) was purchased from Prospect (ENZ-313).
Radioactive filter binding assay
The aptamer, M9-5, was dephosphorylated and labeled with 5′-[γ-32P]-triphosphate as described previously (Ray et al., 2012). The end-labeled RNA was then used in the double-filter nitrocellulose-binding assay. Briefly, the proteins were serially diluted in buffer F (20 mM HEPES pH 7.5, 150 mM NaCl, 2 mM CaCl2, and 3 mM MgCl2), and equal amount of labeled RNA (5,000 cpm, corresponding with a final RNA concentration less than 0.3 pM) were added to each protein dilution at 37°C. The reactions were loaded onto a vacuum manifold containing a nitrocellulose membrane (Whatman) placed over a nylon membrane (Perkin-Elmer). The membranes were exposed to a phosphorimager screen, scanned, and quantitated with a Storm 825 Phosphorimager (GE Healthcare). The corrected fraction bound (FB) was calculated by dividing counts on the nitrocellulose by the total counts and adjusting for background.
Cyclophilin B (CypB) Western blot analysis
Proteins were resolved using 4%–20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) then transferred electrophoretically to polyvinylidene fluoride membranes. The washed membranes were then analyzed by western blotting by using a rabbit polyclonal anti-CypB antibody (1:5,000 dilution; Abcam) with a goat anti rabbit-horseradish peroxidase (HRP) secondary antibody (1:1,000 dilution; Invitrogen). The signal was detected by the Pierce® ECL Western Blotting Substrate (Thermo Scientific).
Glycostaining
5 μg of proteins were resolved by using a 4%–20% SDS-PAGE and subjected to the glycoprotein staining protocol based on the periodic acid-Schiff (PAS) method that detects glycoproteins containing oxidizable glycans (Pierce glycoprotein staining kit, Thermo Scientific).
MiaPaCa-2 secretome preparation
MiaPaCa-2 secretome was purified as described previously (Ray et al., 2012). Briefly, the MiaPaCa-2 cells were grown to 80% confluency in the Dulbecco's modified Eagle medium (DMEM) media containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin antibiotic. Next, media was discarded and the cells were washed in DMEM media (without FBS) and left to grow for another 16 hours in DMEM (without FBS). The conditioned media was collected and concentrated by using the Vivaspin 20 columns (3.0 kDa cutoff, Sartorius Stedim Biotech). The protein concentration of the concentrated media (secretome) was estimated by using Bio-rad protein assay reagent (Bio-Rad) and was stored at −80°C for use in subsequent assays. For PTM analysis, CypB was purified from the MiaPaCa-2 secretome (sample 4) using similar methods as described previously (Ray et al., 2012).
Wheat germ agglutinin-agarose and concanavalin A-agarose column purification
Wheat germ agglutinin (WGA) and concanavalin A (ConA) are lectins that bind to sialic acid/N-acetyl glucosamine-containing and N-linked glycans respectively, and are used to purify glycoproteins. MiaPaCa-2 secretome (400 μl, 0.2 μg/μL) was fractionated by using the WGA-agarose and ConA agarose columns using the Glycoprotein Isolation Kit (Pierce Biotechnology, Thermo Scientific). The bound proteins (eluate) and the unbound fraction (flow-through) were dialyzed against the buffer F in dialysis cassettes (3.5 kDa cutoff, Thermo Scientific) and subjected to the filter-binding assay using radiolabeled M9-5.
Sample preparation and nano-flow liquid chromatography electrospray ionization tandem mass spectrometry analysis
All of the protein samples (in solution) were buffer exchanged into 50 mM ammonium bicarbonate, pH 8.0 using Zebaspin gel-filtration columns (Pierce) and were then subjected to a micro Bradford assay (Pierce). Samples were supplemented with 0.1% Rapigest SF surfactant (Waters Corporation) and then reduced with 5 mM dithiolthreitol for 30 minutes at 70°C and alkylated with 10 mM iodoacetamide for 45 minutes at room temperature. Proteolytic digestion was accomplished by the addition of 500 ng sequencing grade trypsin (Promega) with incubation at 37°C for 18 hours. Supernatants were collected following a 2 minutes centrifugation at 1,000 rpm, acidified to pH 2.5 with trifluoroacetic acid and incubated at 60°C for 1 hour to hydrolyze remaining Rapigest surfactant. Insoluble hydrolyzed surfactant was cleared by centrifugation at 15,000 rpm for 5 minutes. Ten percent of the resulting peptide mixture was then subjected to downstream liquid chromatography electrospray ionization tandem mass spectrometry (LC-MS/MS) analysis.
Each sample was subjected to chromatographic separation on a Waters NanoAquity ultra performance liquid chromatography equipped with a 1.7 μm BEH130 C18 75 μm ID×250 mm reversed-phase column. The mobile phase consisted of (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile. Following a 5-μL injection, peptides were trapped for 5 minutes on a 5 μm Symmetry C18 180 μm ID×20 mm column at 20 μL per minute in 99.9% A. The analytical column was held at 5% B for 5 minutes then switched in-line and a linear elution gradient of 5% B to 40% B was performed over 90 minutes at 400 nL per minute. The analytical column was connected to a fused silica PicoTip emitter (New Objective) with a 10-μm tip orifice and coupled to a Waters Synapt G2 QToF mass spectrometer through an electrospray interface operating in a data-dependent mode of acquisition. The instrument was set to acquire a precursor MS scan from m/z 50-2000 with MS/MS spectra acquired for the three most abundant precursor ions. For all experiments, charge-dependent collision-induced dissociation energy settings were employed and a 120-second dynamic exclusion was employed for previously fragmented precursor ions.
Qualitative identifications and selected ion chromatogram generation from raw LC-MS/MS data
Raw LC-MS/MS data files were processed in Mascot distiller (Matrix Science) and then submitted to independent Mascot searches (Matrix Science) against a SwissProt database (human taxonomy) containing both forward and reverse entries of each protein (20,322 forward entries). Search tolerances were 10 ppm for precursor ions and 0.04 Da for product ions using trypsin specificity with up to two missed cleavages. Carbamidomethylation (+57.0214 Da on C) was set as a fixed modification, whereas oxidation (+15.9949 Da on M), deamidation (+0.98 Da on NQ), and phosphorylation (+79.9663 Da on S, T, and Y) were allowed for phosphopeptide enriched samples. For nonenriched samples, those modifications as well as acetylation (+42.01 Da on K and protein N-terminus), methylation (+14.02 Da on K, R), and ubiquitination (+114.04 Da on K) were allowed. All searched spectra were imported into Scaffold (v4.0, Proteome Software), and scoring thresholds were set to achieve a peptide false discovery rate of less than 1% using the PeptideProphet algorithm (Keller et al., 2002; Nesvizhskii et al., 2003). A scaffold file with all search results is available here: https://discovery.genome.duke.edu/express/resources/3505/Ray_080613_Frozen.sf3
Results and Discussions
Human CypB expressed in mammalian (Human Embryonic Kidney HEK293) cells and in bacteria (E. coli) were assessed for M9-5 binding activity using the radiolabeled filter-binding assay. M9-5 demonstrated binding affinity towards the CypB expressed in HEK293 cells with a mean equilibrium dissociation constant (Kd) of ∼50 nM, whereas it had no significant binding affinity for the bacterially expressed CypB (Fig. 1B). Full-length proteins, with no detectable degradation, were visualized when the proteins were subjected to either western blot or Coomassie staining (Fig. 1A and Fig. 2A, lower panel). CypB (HEK293) has a C-terminal polyhistidine tag, so we used two proteins with polyhistidine tags, PRDX-1 (peroxiredoxin-1) and GRP78 (glucose-regulated protein, 78 kD) as controls. Neither of the control proteins demonstrated significant binding affinity towards M9-5, thus demonstrating that the polyhistidine tag on the CypB (HEK293) is not responsible for the binding affinity (Fig. 1B). PRDX-1 and CypB are basic proteins with calculated isoelectric points (pI) of 8.27 and 9.25 respectively. M9-5, being an RNA aptamer with a negatively charged phosphate backbone, should demonstrate a natural propensity to bind positively charged protein targets. M9-5 showed no significant binding affinity towards PRDX-1 or CypB (produced in E. coli), thus demonstrating that the aptamer–target interaction is not solely dictated by the electrostatic interaction.
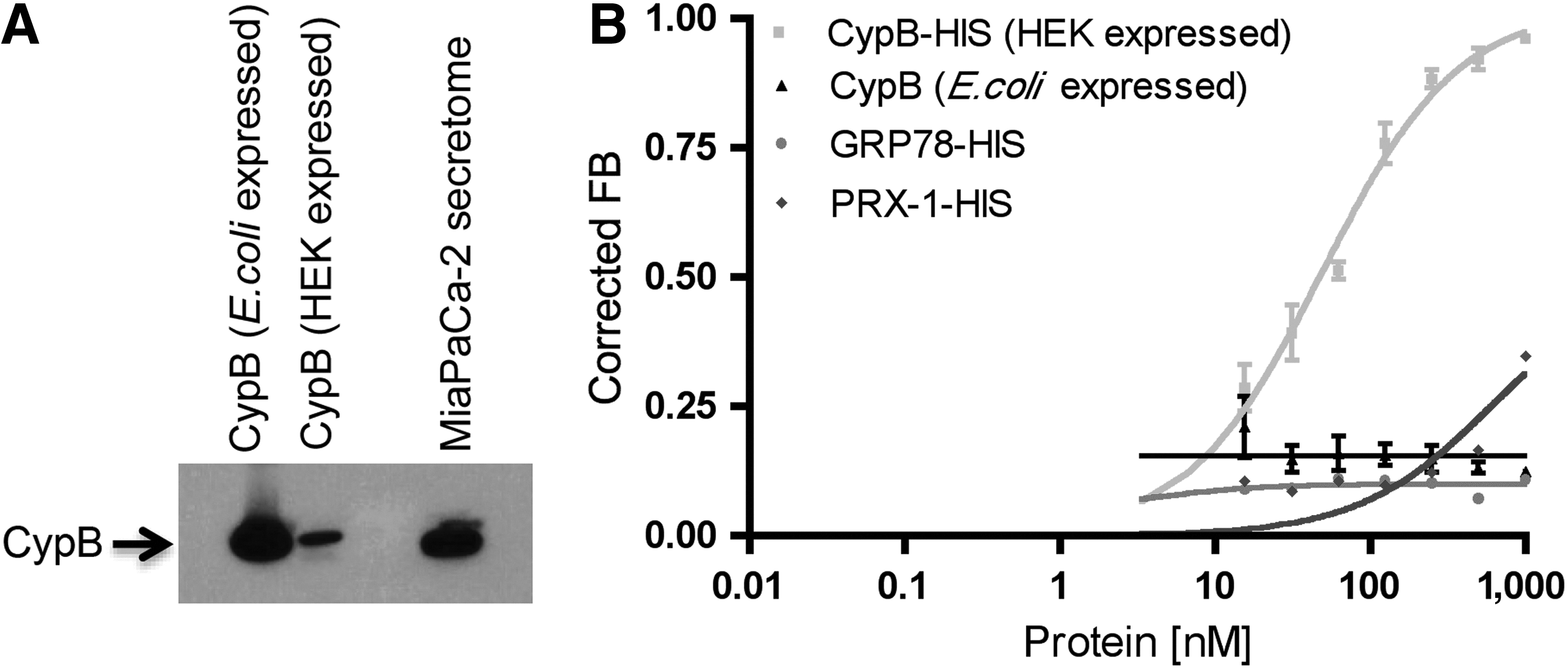
Cyclophilin B aptamer M9-5 directly binds to the purified recombinant cyclophilin B (CypB) expressed in mammalian (HEK293) cells.

Secreted cyclophilin B is non-glycosylated.
Most secreted proteins that are expressed in mammalian cells are glycosylated. Prokaryotes lack the extensive machinery required for glycosylation, and, consequently, proteins expressed in these systems often lack glycan modifications. We hypothesized that the difference in M9-5 binding observed between the human CypB expressed in bacterial versus mammalian cells might be due to lack of glycosylation in CypB protein obtained from the bacterial expression system. To test this hypothesis, we resolved the two proteins—human CypB expressed in bacteria and mammalian cells—in a 4%–20% SDS polyacrylamide gel and subjected them to a glycostaining protocol based on the PAS method. Briefly, the cis-diol groups of saccharide present in the glycoproteins are initially oxidized to aldehydes in the presence of the periodate-based oxidizing agent. The resulting aldehyde groups then react with the glycostaining reagent to form Schiff-base bonds that form magenta polypeptide bands with either light pink or a colorless background. HRP, a heavily gylcosylated protein, and soybean trypsin inhibitor, a non-glycosylated protein, were used as positive and negative controls, respectively. No magenta-colored bands were observed with CypB expressed in either the bacterial or mammalian cells, indicating that the proteins were non-glycosylated (Fig. 2A, upper panel). Proteins with glycan modifications have higher molecular weight than their non-glycosylated versions due to the added glycan groups. Due to this, the glycosylated proteins tend to migrate more slowly than their non-glycosylated forms in SDS-PAGE and can be detected as higher migrating bands when subjected to Coomassie staining or western blot analysis. However, in the case of CypB, the proteins expressed in both bacterial and mammalian cells demonstrated similar migration patterns [Coomassie staining (Fig. 2A, lower panel) and western blots analysis (Fig. 1A)].
Wheat germ agglutinin (WGA) and Concanavalin A (ConA) are lectins that bind glycans and are used to purify glycoproteins. We fractionated the MiaPaCa-2 secretome using WGA-agarose and ConA-agarose columns and analyzed the protein fractions that were bound to the columns (eluate) and the unbound fractions (flow-through) for the presence of CypB (Fig. 2B). The rationale was that if CypB was glycosylated then it should bind to these columns and would be present in the eluate. First, as a quality control step, we subjected the flow-through and eluate from the WGA and ConA columns to silver staining to check the fractions for the presence of proteins (Fig. 2C). The fractions were next subjected to western blot analysis using anti-CypB antibody. Cyp B was detected in the flow-through but not the eluate fractions of both the WGA and ConA columns indicating that CypB did not bind to these lectin columns (Fig. 2D). Consistent with these findings, when the M9-5 binding activity was tested using the radiolabeled M9-5 filter-binding assay, we found that the binding activity was present in the flow-through fractions and not in the eluate fractions from these columns (Fig. 2E). Taken together, these data strongly suggest that CypB from the MiaPaCa-2 secretome is non-glycosylated. However, the possibility exists that there might be minor glycan modifications that are not detected by the glycoprotein staining method and do not result in binding to the lectin columns that were used. The slight increase in the molecular weight resulting from a minor glycosylation of CypB might not be significant enough to be detected as a differential protein migration pattern in SDS-PAGE.
The result was unexpected, as most secreted proteins in mammalian systems are known to be glycosylated. However, there are other PTMs—such as phosphorylation, acetylation, ubiquitination, methylation, deamidation, and oxidation—that might explain the M9-5 specificity for CypB expressed by mammalian cells. To investigate these PTMs further, we subjected CypB from different sources to mass-spectrometric analysis. Samples were subjected to an unbiased LC-MS/MS analysis to map various PTMs. All data were searched against a SwissProt database (human taxonomy) and allowed for the possibility of the following PTMs: phosphorylation, acetylation, ubiquitination, methylation, deamidation. and oxidation. We analyzed three recombinant CypB samples produced in mammalian cells (samples 1, 2 and 3), endogenous CypB purified from human pancreatic cancer cells (sample 4) (Fig. 3), and recombinant CypB produces in bacteria (sample 5) independently and cumulatively across all the samples, and a number of high confidence PTMs were mapped (Table 1).
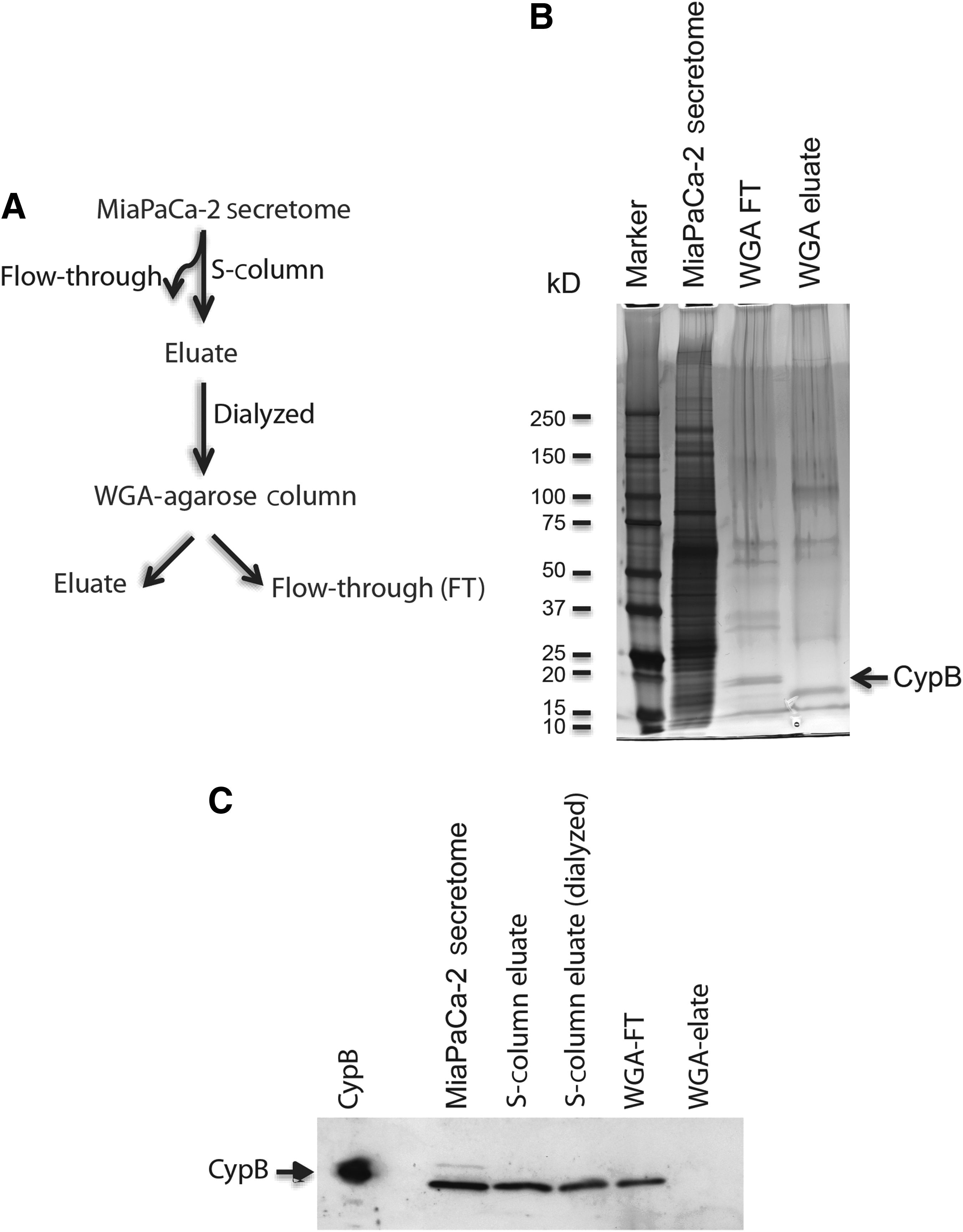
Purification of CypB from the MiaPaCa-2 secretome for the mass-spectrometric analysis and posttranslational modification search.
Samples 1, 2, and 3 are recombinant human CypB expressed in HEK293 cells; sample 4 is the CypB purified from the MiaPaCa-2 secretome (Fig. 3); and sample 5 is recombinant human CypB expressed in Escherichia. coli cells. Data were annotated with a maximal 1% peptide false discovery rate (FDR) using the Peptide/Protein Prophet algorithm within Scaffold, but the actual measured FDR was 0%. Amino-acid abbreviations: K=lysine; T=threonine; S=serine. The posttranslational modifications that were identified more than once in different samples are highlighted in boldface.
A scaffold file with all search results is available online at: https://discovery.genome.duke.edu/express/resources/3505/ray_080613_frozen.sf3
Both mammalian-expressed recombinant CypB and endogenous purified CypB were acetylated at lysine residue K98. Cyclophilin A (CypA), the isozyme of CypB, is also acetylated in human cells, and Lammers et al. demonstrated that acetylation regulates the functions of CypA in immunity and viral infection (Lammers et al., 2010). Additionally, mammalian-expressed recombinant CypB was ubiquitinylated at lysine residues K67, K71, K84, K98, K158, K165, K192, K195, and K204. Proteins are generally tagged with poly-ubiquitin in the cytoplasm for proteasome-mediated degradation (RECHSTEINER, 1987; WILKINSON, 1987). However, recent studies have demonstrated that ubiquitin can attach to target proteins by eight different linkages, and the linkage type can dictate whether a protein would be degraded, bound to other proteins to initiate a signaling cascade, or endocytosed in case of membrane-bound receptors (KOMANDER, 2009; Strieter and Korasick, 2012). It is unusual for a secreted protein, such as CypB to be ubiquitinylated. However, there are examples of secreted proteins being ubiquitinylated, and there are ubiquitination enzymes present in the extracellular compartment (Baska et al., 2008; HuangFu et al., 2010). CypB was also found to be ubiquitinylated in a whole proteome wide study for ubiquitin-modified proteins (Kim et al., 2011; Wagner et al., 2011). Methylation at lysine K71 and phosphorylation at threonine T72 were also detected in the CypB PTM analysis.
In contrast, the bacterially produced CypB was acetylated at 16 lysine residues (K67, K71, K84, K98, K116, K129, K131, K145, K158, K165, K181, K186, K192, K195, K204, and K215) (Table 1, sample 5). Recent studies have suggested that acetylation of proteins at lysine residues in bacterial systems is a more widespread PTM than previously thought and might have regulatory roles in their metabolic pathways (Yu et al., 2008; Weinert et al., 2013; Zhang et al., 2013). Acetylation of lysine side chains is associated with the neutralization of positive charge and has been extensively studied in the context of histone–DNA interaction. The acetylation of the ɛ-amino group of lysine residues in the N-terminal tail of histones neutralizes the positive charge of the protein and causes the DNA to unwrap and thus adapt a more open nucleosome conformation during gene transcription (STRUHL, 1998; Shahbazian and Grunstein, 2007). The same lysine residues (K67, K71, K84, K98, K158, K165, K192, K195, and K204) that were ubiquitinylated in the mammalian-expressed CypB were acetylated in the bacterially expressed CypB (Table 1) as were several other lysine residues. It is our hypothesis that the differential ubiquitinylation and acetylation of lysine residues, with a net neutralization of positive charge in the heavily acetylated bacterially expressed protein, is the mechanism behind the selectivity of M9-5 for the mammalian-expressed CypB. However, since there are other PTM differences between the two proteins, it is possible that simple change in electrostatic interaction is not the sole mechanism for this selectivity. Regardless, these observations reinforce the importance of using proteins expressed in mammalian cells for SELEX if the intended application for the resultant aptamers is for mammals.
The biological implications of the PTMs on CypB described here are unknown. The M9-5 aptamer, which can differentiate the sera of patients with pancreatic cancer from healthy volunteer sera (Ray et al., 2012), can also distinguish between the human CypB expressed in mammalian cells versus bacteria. In contrast, the CypB antibodies we tested did not distinguish between the two forms of CypB. Further biochemical and structural confirmation is required to determine whether and which of the PTMs identified in this study are responsible for the aptamer selectivity. Site-directed mutagenesis could be utilized to evaluate the contribution of PTMs on specific amino acids, individually or in combination, but the large number of candidate PTMs would make this approach impractical. Additionally, these mutations may also disrupt the structural integrity of the protein and influence the aptamer–protein interaction directly. In our opinion, the best way to address the question of the role of PTMs would come from solving the co-crystal structure of the aptamer with CypB. While far from trivial, this approach would directly indicate which amino acids are involved and whether there are PTMs involved in the aptamer–CypB interaction. Meanwhile, we have not yet proven that this selectivity confers improved performance for this particular biomarker. However, given the roles that PTMs have been shown to play as regulators and markers of disease, the ability to distinguish between forms of the same protein with differing PTMs is an important advantage of aptamers as tools for identification and detection of biomarkers.
Footnotes
Acknowledgments
We thank Meredith Turner, Erik Soderblom, and Arthur Moseley in Duke University's Proteomics Core Facility for the mass spectrometry analysis. This work was supported by the National Institutes of Health under award no. K08CA142903 (RRW) and the Duke Cancer Institute.
Author Disclosure Statement
No competing financial interests exist.