
Abstract
Targeting of pre-mRNA by short splice-switching oligonucleotides (SSOs) is increasingly being used as a therapeutic modality, one rationale being to disrupt splicing so as to remove exons containing premature termination codons, or to restore the translation reading frame around out-of-frame deletion mutations. The aim of this study was to investigate the effect of chemically linking individual SSOs so as to ascertain equimolar cellular uptake that would provide for more defined drug formulations. In contrast to conventional bispecific SSOs generated by conjugation in solution, here we describe a protocol for synthesis of bispecific SSOs on solid phase. These SSOs comprised of either a non-cleavable hydrocarbon linker or disulfide-based cleavable linkers. To assess the efficacy of these SSOs we have utilized splice switching to bypass a disease-causing mutation in the DMD gene concurrent with disruption of the reading frame of the myostatin gene (Mstn). The premise of this approach is that disruption of myostatin expression is known to induce muscle hypertrophy and so for Duchenne muscular dystrophy (DMD) could be expected to have a better outcome than dystrophin restoration alone. All tested SSOs mediated simultaneous robust exon removal from mature Dmd and Mstn transcripts in myotubes. Our results also demonstrate that using cleavable SSOs is preferred over the non-cleavable counterparts and that these are equally efficient at inducing exon skipping as cocktails of monospecific versions. In conclusion, we have developed a protocol for solid-phase synthesis of single molecule cleavable bispecific SSOs that can be efficiently exploited for targeting of multiple RNA transcripts.
Introduction
A
DMD is a common, fatal muscle degenerative disorder affecting approximately 1 in 3,500 newborn boys. The disease is caused by mutations in the DMD gene that typically disrupt the open reading frame of the dystrophin mRNA leading to absence of functional protein (Hoffman et al., 1987). The existence of mildly affected Becker muscular dystrophy individuals with in-frame DMD gene deletions suggests that modification of dystrophin pre-mRNA splicing by forced skipping of appropriate exons, using SSOs, could induce the production of partly functional dystrophin in DMD patients (Aartsma-Rus et al., 2003). Indeed, the use of SSOs to mediate such splice switching therapy has been demonstrated numerous times in vivo using the dystrophin-deficient mdx mouse model (Lu et al., 2003, 2005; Alter et al., 2006; Wu et al., 2008, 2010, 2012; Heemskerk et al., 2009; Yin et al., 2010). The therapeutic potential of this strategy has recently been shown in two independent systemic clinical trials. In one of the studies the authors showed dose-dependent SSO efficacy, using the AON analog 2′ -O-methyl phosphorothioate RNA (2′ -OMePS RNA), in patients with DMD (Goemans et al., 2011). Similarly, a phase 2 systemic trial using phosphorodiamidate morpholino oligomer (PMO) chemistry has also demonstrated dose dependent restoration of dystrophin, albeit at low levels (Cirak et al., 2011).
Although this shows that SSOs have therapeutic potential, targeting single mutations in DMD with SSOs can only benefit a fraction of patients suffering from the disease since different mutations require different SSOs. Extensive efforts have therefore been made to develop tools to skip multiple exons (i.e., Ex45–55) in DMD patients that would allow a single treatment to address almost 75% of DMD patients suffering from deletion mutations (van Vliet et al., 2008). Furthermore, several other proteins could contribute to the pathology of DMD and targeting several pathways simultaneously may therefore be beneficial. One important consideration has been to devise an efficient means of assuring equal delivery of multiple SSOs into the same cells. Researchers have attempted to induce multi-exon skipping of the large deletion mutation “hotspot” regions of the DMD gene (exons 45–55) using cocktails of SSOs with limited success (van Vliet et al., 2008), although it should be noted that skipping with two SSOs to correct DMD mutations has been successfully demonstrated (Aartsma-Rus et al., 2004; McClorey et al., 2006). Apart from DMD, others have developed so-called bispecific AONs to simultaneously target different mRNAs with the same AON (Rubenstein et al., 2012). Most of these bispecifics have been constructed to contain two antisense sequences spaced by nucleotide hairpins and can therefore only target either one of two sites. In 2010, the first bispecific short interfering RNA (siRNA) was reported in which two siRNAs with different cellular targets were connected together by a cleavable disulfide linker (Mok et al., 2010). The authors demonstrated that such a single molecule siRNA was cleaved inside the reductive environment of cells and could mediate robust RNA interference (RNAi) responses for both target genes. However, these bispecific siRNAs were generated by mixing separate siRNAs harboring free thiol groups which resulted in mixtures of monospecific, bispecific and multimeric siRNAs (and were thus not of a defined composition). Follow-up work from the same group showed that defined cleavable bispecific siRNAs could be generated by covalent conjugation in solution (Chung et al., 2011). However, the yields of such reactions are typically relatively low.
Here, we sought to develop single molecule bispecific SSOs that are synthesized with internal cleavable linkers (i.e., disulfide bridges) on solid phase (Fig. 1) and that potentially can be extended with further antisense sequences for multiple targeting. In the context of DMD, studies have primarily been based on targeting removal of a single exon within Dmd pre-mRNA. However, other proteins and small regulatory microRNAs (miRNAs) have recently been shown to be involved in the pathology of DMD (McPherron et al., 1997; Cacchiarelli et al., 2011; Roberts et al., 2012b) and are therefore potential therapeutic targets. Of particular interest is myostatin (Mstn), a secreted transforming growth factor beta (TGFβ) superfamily member, which has a role in muscle homeostasis and is thought to play a major role in various muscle wasting disorders (McPherron et al., 1997). In mouse models of DMD, the simultaneous blockade of the myostatin pathway leads to muscle hypertrophy and concurrent with dystrophin restoration has been suggested to have a greater therapeutic outcome than dystrophin restoration alone (Bogdanovich et al., 2002).
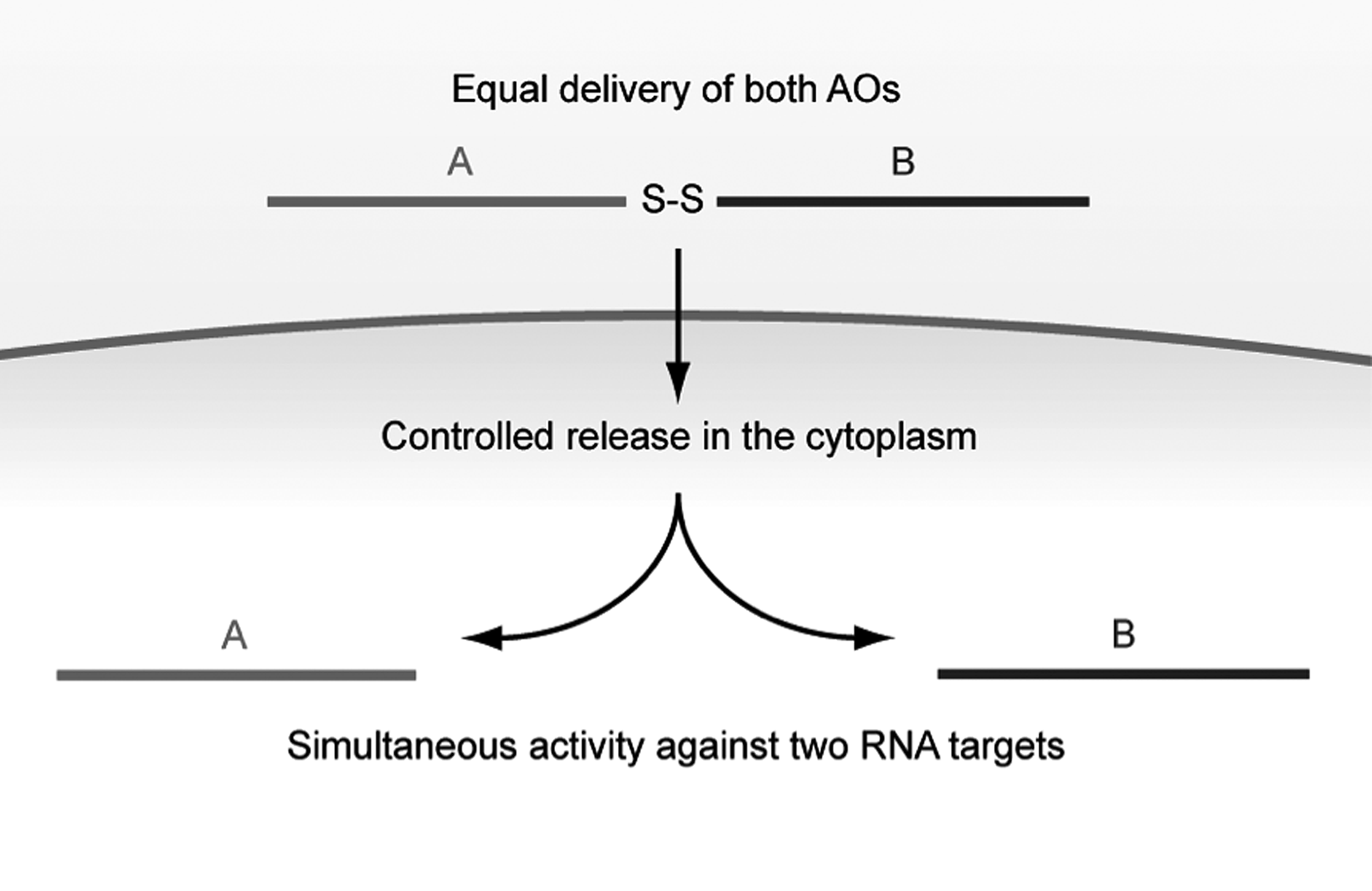
Principle of single molecule bispecific short splice-switching oligonucleotides (SSOs) with internal cleavable disulfide linkers. Two SSO sequences (
Thus, we here synthesized bispecific SSOs where one arm targets exon 23 of the mouse Dmd locus and the other targets exon 2 of the Mstn gene to disrupt the reading frame, as previously reported by others (Kang et al., 2011b; Kemaladewi et al., 2011; Malerba et al., 2012). These bispecific oligonucleotides were either synthesized having internal disulfides, or with non-cleavable C18 spacers and then compared with the use of singular SSOs. Our results demonstrate that bispecific SSOs based on 2′-OMePS RNA chemistry can be readily synthesized on solid phase with high yields and purity. Importantly, such SSOs could effectively promote exon skipping of both transcripts in differentiated H2k mdx myotubes. As expected, the cleavable bispecific SSOs displayed greater activity than the non-cleavable version, being as potent as, or even better, than a cocktail of monospecific SSOs.
Materials and Methods
Synthesis of oligonucleotides
All oligonucleotide derivatives were synthesized on an ÄKTA™ oligopilot™ 10 plus system at GE Healthcare Bio-Sciences in Uppsala, Sweden. Since priority was given to purity and that these oligonucleotide derivatives were ∼45–50 nucleotides in length, loading was lowered and an extra spacer was used to facilitate synthesis performance. Standard template 2′ -OMe RNA synthesis methods were used except thiolation which was carried out with a 1:1 mixture of acetonitrile (ACN) and PADS [(bis(diphenylacetyl)disulfide), ISIS Pharmaceuticals, 0.2 M in ACN/3-picoline 1:1(v/v)]. Introduction of spacer (C18) and linker (SS or XS) were carried out by double couplings (0.1 M amidite, 4 equiv.), 2×10 minutes followed by standard oxidation [50 mM I2, Pyr/H2O 9:1 (v/v) in a 1:1 mixture of ACN]. Dilution of oxidation/thiolation reagent is to avoid reductive cleavage of the disulfide. Other ancillary reagents (EMD Chemicals) were used as recommended by GE Healthcare Bio-Sciences. The C18 (spacer phosphoramidite 18 No. 10-1918-xx), SS (thiol modifier C6 Phosphoramidite # 10-1936-xx), and XS (dithiol phosphoramidite, DTPA, # 10-1937-xx) were purchased from Glen Research and used as recommended by the supplier. Cy5 amidite (GE Healthcare No. 28904249) was used as recommended by supplier. Final detritylation and dietylamine treatment was done (to remove beta-cyanoethyl groups) prior to cleavage and deprotection overnight in 25% aqueous ammonium hydroxide (Merck) at 55°C, releasing the crude oligonucleotide derivative into solution ready for purification.
Purification and desalting of SSOs
All purification was carried out using an ÄKTAexplorer™ 100 system equipped with Capto™ Q ImPres columns for anion-exchange chromatography (AEC) (buffer A: 10% ACN, 10 mM NaClO4, 50 mM TRIS, pH 7.5; 1mM EDTA; buffer B: 10% ACN, 500 mM NaClO4, 50 mM TRIS, pH 7.5; 1 mM EDTA). The ÄKTAexplorer system comprises a P-900 high-performance liquid chromatography (HPLC) pump, a P-960 sample pump, a model UV-900 monitor model pH/C-900 pH/conductivity monitor and the system is controlled with UNICORN™ software.
After AEC purification, the SSOs were in a solution containing sodium perchlorate. The salt was removed from the samples by gel filtration with an isocratic flow (3 mL/minute) of Milli-Q™ water. Five HiTrap™ desalting columns were mounted in serial on the ÄKTAexplorer system and enabled desalting of one complete 12 mL fraction in about 15 minutes.
Analysis and characterization of SSOs
Analytical HPLC was used with 15 mM triethylamine/ 400mM hexafluoroisopropanol in water as buffer A and methanol as buffer B. Liquid chromatography–mass spectrometry (LC-MS) was performed using an Ettan™ LC (GE Healthcare Bio-Sciences) connected to a Finnigan™ LCQ™ Deca XP plus mass spectrometer (ThermoFischer Scientific), for the characterization of individual fractions (e.g., Dys-SS-Myo) after purification and desalting.
Xevo™ G2 QTof together with an AQUITY™ UPLC H-class (Waters Sweden AB) equipped with a ACQUITY UPLC OST C18, 1.7 μm, 2.1×50 mm column controlled by MassLynx™ was used for purity analysis and characterization of most oligonucleotide derivatives (e.g., Myo-XS-Dys) together with Maxent1 software for molecular weight calculation (Waters Sweden AB).
Cell culture and transfection of cells
Mouse H2k mdx (Morgan et al., 1994) muscle cells were grown on gelatinized plates at 33°C, 10% CO2 in Dulbecco's modified Eagle's medium (DMEM) with glutamax supplemented with 20% fetal bovine serum, 0.5% chick embryo extract, 200 U/mL penicillin and 200 μg/mL of streptomycin (Invitrogen). H2k mdx myotubes were generated in gelatin-coated 24-well plates by seeding 30,000 cells per well, leaving them for 2 days to reach 90% confluency before changing media to starvation media (DMEM supplemented with 5% horse serum) and transferring them to 37°C, 5% CO2 incubator for another 4 days. Cells were transfected using Lipofectamine2000 (LF2000; Invitrogen) according to manufacturer's protocol. Briefly, 2.2 μL of LF2000 was used per μg of AON. Complexes were formed in 50 μL OptiMEM (Invitrogen) and added to cells grown in 450 μL full growth media. Cells were processed 48 hours later in all transfection experiments.
Animal experiments
Experiments were carried out in the Biomedical Sciences Unit, University of Oxford according to procedures authorized by the UK Home Office. Intramuscular injections of AONs into the tibialis anterior were carried out on 7–9 week old C57BL/10ScSn-Dmdmdx/J mdx mice under isoflurane anesthesia. Briefly, 10 μg of AON was formulated with InVivo jetPEI (PolyPlus Transfection) at an N/P ratio of 10 in 10% glucose, according to manufacturer's instructions. Mice were sacrificed seven days later and muscles were snap-frozen in liquid nitrogen and stored at –80°C.
RNA extraction and reverse transcription–polymerase chain reaction
RNA was extracted from either H2k mdx monolayer cultures or 50×8 μm frozen muscle sections using Trizol (Invitrogen) according to manufacturer's instructions. In order to assess the degree of exon skipping, 400 ng of total RNA was used as a template in a 50 μL reverse transcription–polymerase chain reaction (RT-PCR) using the GeneAmp RNA PCR kit (Applied Biosystems). Nested RT-PCR analysis of the Dmd transcript between exons 20 to 26 was performed under the following conditions; 95°C for 20 seconds, 58°C for 60 seconds, and 72°C for 120 seconds for 30 cycles, with 2 μL of this reaction used as a template for nested amplification using Amplitaq Gold (Applied Biosystems) under the following conditions; 95°C for 20 seconds, 58°C for 60 seconds, and 72°C for 120 seconds for 25 cycles. For myostatin transcript analysis, nested amplification of exons 1–3 was performed under the following conditions; 95°C for 20 seconds, 56°C for 30 seconds and 72°C for 60 seconds for 35 cycles with 2 μL of this reaction used as a template for nested amplification; 95°C for 20 seconds, 56°C for 30 seconds, and 72°C for 60 seconds for 25 cycles. PCR products were analyzed on 2% agarose gels and densitometry performed by QuantityOne software (BioRad). Exon 23 skipping levels are expressed in the text as the percentage of the of exon 23 skipped PCR product intensity relative to the sum of the intensities of the exon 23 skipped and full-length products. Similarly, Mstn exon 2 skipping is expressed as the percentage of the of exon 2 skipped product intensity relative to the sum of the intensities of full-length and exon 2 skipped products. Additionally, to quantify levels of Mstn exon 2 skipping in vivo, 1 μg of RNA was reverse transcribed using the High Capacity cDNA RT Kit (Applied Biosystems) according to manufacturer's instructions. Quantitative polymerase chain reaction (RT-qPCR) analysis was performed using 25 ng cDNA template and amplified with TaqMan Gene Expression Master Mix (Applied Biosystems) on a StepOne Plus Thermocycler (Applied Biosystems). Levels of Mstn exon 2 skipping were determined by RT-qPCR of exon 2–3 (Assay Mm.PT.56a.13573446, Integrated DNA Technologies), normalized to Cyclophilin B (Assay Mm00478295, Applied Biosystems). Primer sequences are available upon request.
Wst-1 cell viability assay
Cell viability was assessed by the Wst-1 proliferation assay according to manufacturer's protocol (Roche Diagnostics Scandinavia AB). Briefly, cells were grown and treated as described above but in 96-well format (i.e., 8,000 cells seeded and treated in 100-μL volume). Two days after transfection at 200 nM SSOs complexed with LF2000, Wst-1 was added to each well. Wst-1 measures the activity of mitochondrial dehydrogenases to convert tetrazolium salts to formazan, and cell proliferation is directly correlated to the amount of formazan product that is formed. Absorbance was measured on SpectraMax Absorbance Microplate reader (Molecular Devices).
Fluorescence-activated cell sorting analysis
H2k mdx myotubes were transfected with LF2000 (Invitrogen, Sweden) according to manufacturer's protocol. After 4 hours, the cells were trypsinized for 10 minutes. After trypsinization the cells were washed twice with DPBS (Dulbecco's phosphate-buffered saline). Before the analysis with flow cytometry, the cells were resuspended in DPBS containing 1 μL/mL propidium iodide (Life Technologies). Fluorescence-activated flow cytometry (FACS) was performed with BD FACS Calibur Flow Cytometry System (Center for Hematology and Regenerative Medicine [HERM], Karolinska Institutet). The analysis of the data was done with FlowJo (Department of HERM, Karolinska Institutet).
DTT cleavage assay
Dried AONs were resuspended in 100 mM 1,4-dithiothreitol (DTT) solution in sodium phosphate buffer (pH 8.3–8.5). The tubes were kept in 37°C overnight. The samples were run on 15% Tris/Borate/EDTA (TBE) urea gel (Life Technologies). After 10 minutes incubation with SYBR Gold Nucleic Acid Gel Stain (Invitrogen), the detection of the gel was performed by VersaDoc system (BioRad) and analysis done using the QuantityOne software (BioRad).
Results
Evaluation of SSOs for myostatin corruptive exon skipping
Before initiating the synthesis of novel bispecific SSOs targeting different transcripts, we first set out to screen for potent SSO sequences targeting the Mstn transcript. Here, we designed SSOs that promoted skipping of exon 2 from the Mstn pre-mRNA to disrupt the mRNA reading frame so as to reduce levels of myostatin protein. Four 25mer AONs were designed to target either the consensus splice sites of exon 2 (AON 1 and AON 4) or putative exon splicing enhancer regions (AON 2 and AON 3) identified using the enhancer prediction algorithms ESEFinder 3.0 (Smith et al., 2006) and RescueESE (Fairbrother et al., 2002) (Fig. 2). Each AON was transfected at 100 nM into H2k mdx cells along with a previously published AON as a positive (+ve) control targetting Mstn (Kang et al., 2011a) (+veAON) or a 25mer AON targeting exon 23 of the Dmd gene (DYS AON) (Fig. 2). All AONs targeting Mstn effectively spliced exon 2 from the mature transcript, with further titration studies demonstrating AON3 to have the greatest efficacy (data not shown). Based on the efficacy of AON3 being as good, if not better than a previously published AON sequence used to successfully induce a functional change in vivo, (Kang et al., 2011a) we considered this appropriately effective for Mstn targeting. This AON sequence (5′-CCAUUCUCAUCCAAAGCUUU GAUUU-3′), that targets positions 304–328 of exon 2, was therefore chosen as the basis for bispecific SSO design.

Development of SSOs for Mstn exon 2 skipping. Four SSOs were developed to target consensus splice sites and exon splicing enhancer regions of exon 2 of the myostatin (Mstn) gene as depicted in schematic. Exon skipping efficacy was determined by nested reverse transcription–polymerase chain reaction (RT-PCR) with 158-bp product representing removal of exon 2 from the mature Mstn transcript. All SSOs successfully removed exon 2, at a similar efficacy to a previously published SSO (+ve). SSOs targeting Dmd gene (exon 23) and untreated cells were used as negative controls. AON, antisense oligonucleotide.
Design of bispecific SSOs
Given the success with cleavable bispecific siRNAs and the limited success with nucleotide-spaced bispecifics for multiexon skipping in DMD (Aartsma-Rus, personal communication), we set out to develop a system to generate single AONs internally spaced with cleavable (i.e., disulfide) linkers or non-cleavable C18 spacers by solid phase synthesis in ÄKTA oligopilot 10 plus system. The synthesis scheme is depicted in Fig. 3. Briefly, standard template 2′-OMe RNA synthesis methods were used, except for thiolation/oxidation conditions (diluted with 50% ACN-as recommended by the supplier (Glen Research) to avoid reductive cleavage of the disulfide linkage). In the case of C18, we used the same synthesis conditions (as for XS and SS) to have a relevant reference synthesis. SSO sequences and nomenclature are listed in Table 1.

Generation of bispecific SSOs.
Bispecific SSOs promote exon skipping of dystrophin and myostatin
After confirmation that we could generate the correct SSO products with high purity (an example is depicted in Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat), we assessed the exon skipping efficacy of either (a) cotransfecting Dmd targeting Ex23D (+7-18) SSO (which has previously been identified as being highly effective; Mann et al., 2002) together with Mstn targeting Ex2A(+304+328) (M+D); (b) a non-cleavable bispecific SSO comprising both sequences spaced by C18 (BI-C18); or (c) a cleavable version internally spaced by a conventional disulfide (BI-S), in H2k mdx myotubes. The efficacy of target exon removal was determined by gel densitometry of nested RT-PCR products as described in Materials and Methods section. As seen in Fig. 4a, all SSOs induced Dmd exon 23 skipping in a dose-dependent manner as determined by RT-PCR analysis. Interestingly, the activity of BI-S was similar or slightly higher as compared to transfection with the separate SSOs and exceeded the activity of the non-cleavable BI-C18. Additionally, all SSOs simultaneously mediated exon 2 removal from the mature Mstn transcript. As we observed near complete exon skipping of the Mstn transcript at 100 nM concentration (Fig. 2), we chose to only assess exon skipping at a lower SSO concentration (50 nM). In contrast to dystrophin, no major differences were observed between the different bispecifics or monospecific cocktails. A summary of independent transfection experiments are provided in Fig. 4b (Dmd) and Fig. 4c (Mstn). These results are in accordance with previous reports using multimeric siRNAs; a single SSO with more than one target site can target several transcripts simultaneously with equal or better efficiency than the separate SSOs (Mok et al., 2010). Unsurprisingly, our results also suggest that a cleavable linker within the bispecific SSOs is preferable over a non-cleavable linker.

Bispecific SSOs efficiently induce exon skipping of both Dmd and Mstn.
We next validated whether such cleavable bispecific SSOs could induce exon skipping in vivo. Eight week-old mdx mice were injected intramuscularly into the tibialis anterior with either monospecific SSOs (5 μg of each) or 10 μg BI-S complexed with InVivo-JetPEI at an N/P ratio of 10. Tissue was harvested 7 days later and RNA analyzed for Dmd Ex23 skipping by RT-PCR and Mstn exon 2 skipping by RT-qPCR. Efficacy of Mstn exon 2 skipping was indirectly assessed by quantitative measurement of Mstn transcripts containing exon 2 (using primers across exon 2–3 boundary) after normalization to housekeeping gene. At these dosage levels, we were unable to detect any dystrophin exon skipping with any of the SSOs (data not shown), although a robust reduction in Mstn transcript levels containing exon 2 could be observed (Fig. 4d). Similar to in vitro transfection results, no significant difference could be observed between BI-S SSO treatments compared with monospecific versions.
The uptake and toxicity profile of bispecific SSOs is similar to the monospecific versions
We have previously observed that the transfection efficiency of nucleic acids using LF2000 may depend on the molecular weight of the cargo (unpublished data
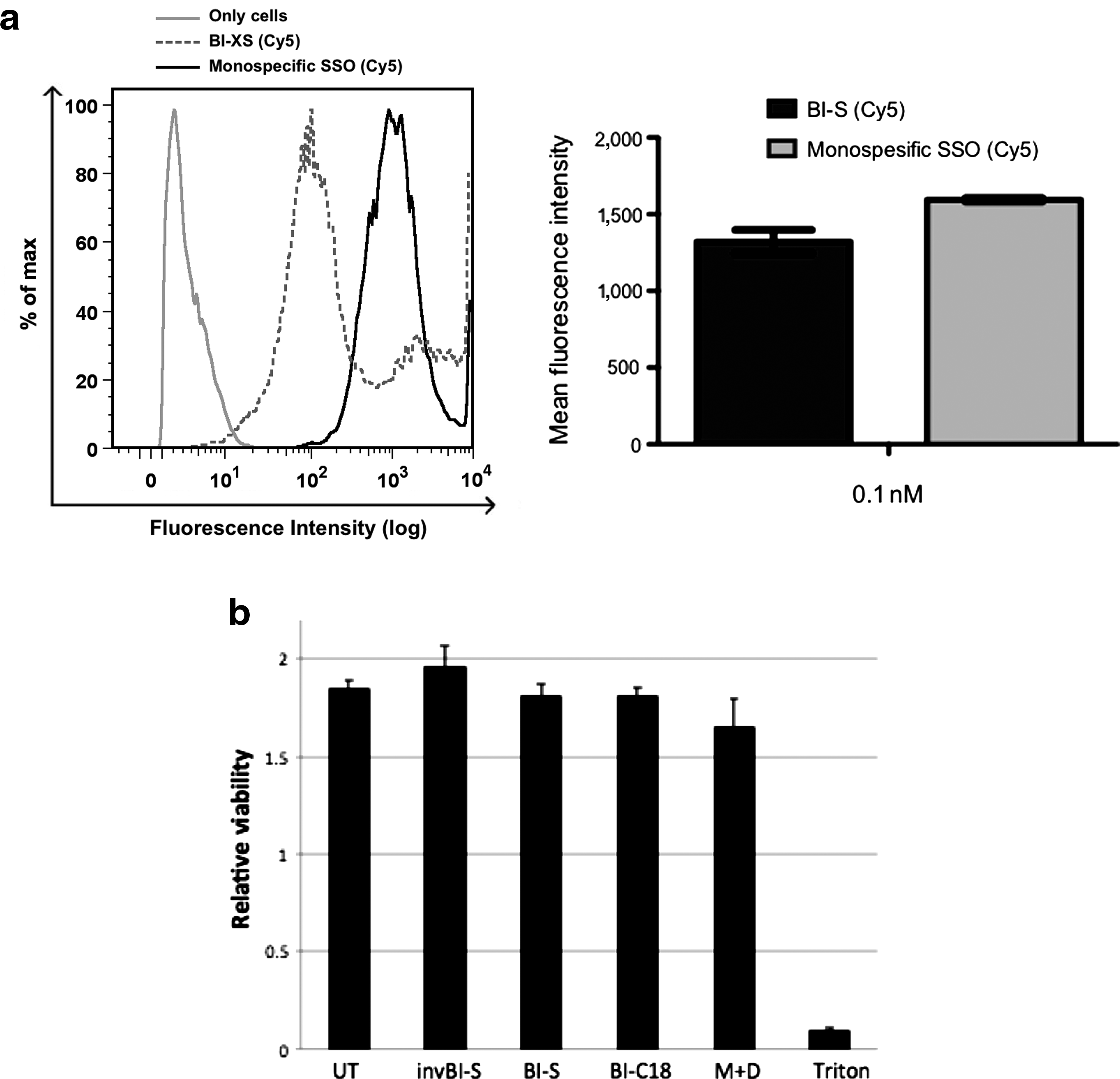
Uptake and toxicity profiles of the different SSOs.
Next, we validated whether the high activity of BI-S was associated with any cellular toxicity. Cells were transfected at 200 nM SSOs and assessed for viability 48 hours later using the Wst-1 assay. None of the treatments showed any toxicity compared to untreated cells whereas the positive control (0.1% Triton in PBS) reduced the viability dramatically (Fig. 5b).
Evaluation of different types of bispecifics
After confirming the activity of BI-S, we next set out to test whether the internal orientation of the antisense sequences (Myo-Dys or Dys-Myo) altered the splice-switching activity. For this, we synthesized yet another version (depicted invBI-S) that contains the Mstn antisense sequence in the 5′ end of the bispecific SSO. Cells were transfected at 50 nM of both bispecific versions and exon skipping assessed 48 hours later. As seen in Fig. 6a, both SSOs mediate exon 23 skipping from the Dmd transcript with similar efficacy. Although invBI-S appeared slightly more active for exon 2 skipping of the Mstn transcript, large variation between experiments make it difficult to conclude if there was a significant difference and at most we can say that internal orientation did not significantly impact on overall activity (Fig. 6a).
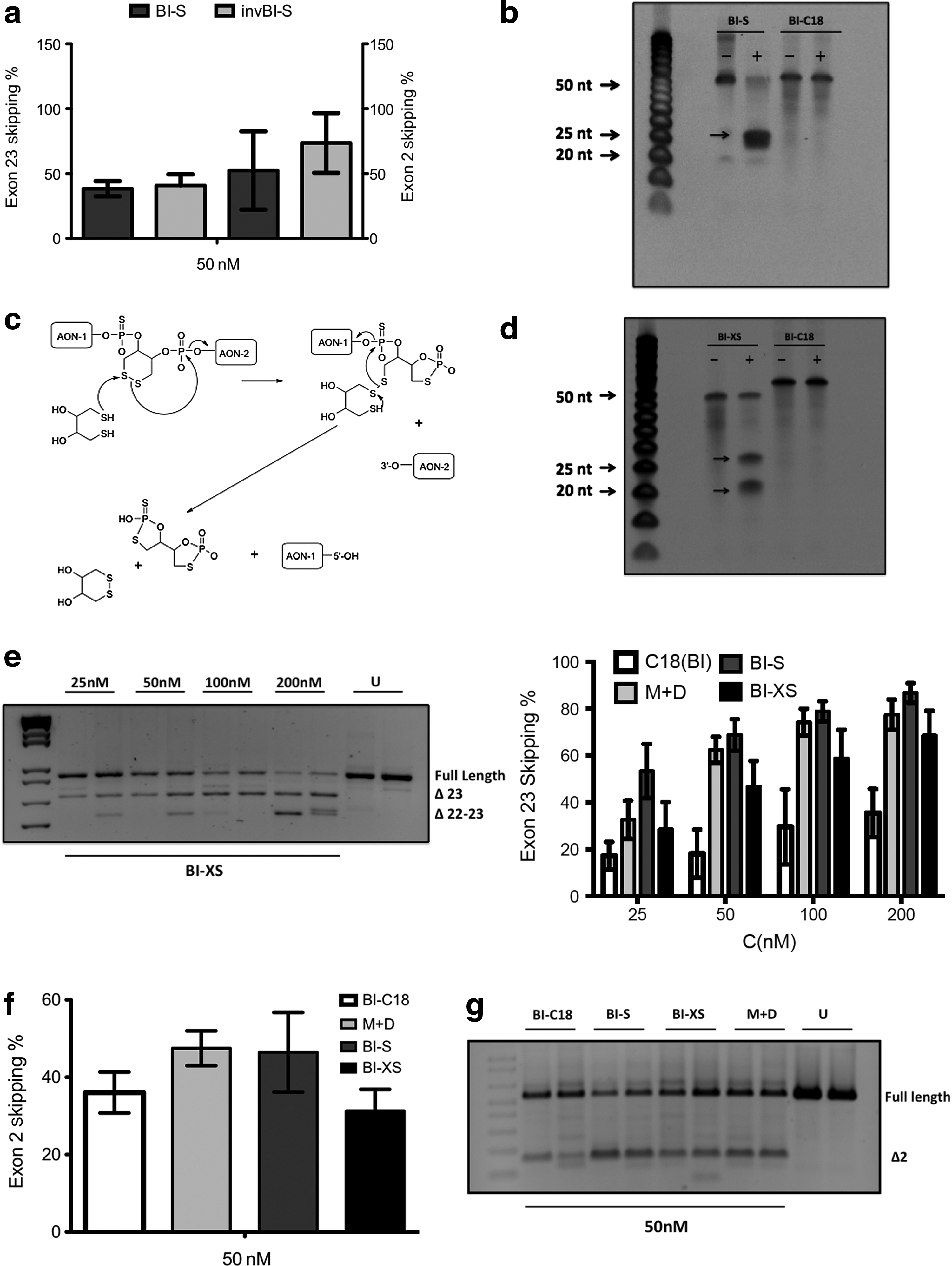
Assessment of exon skipping activity and stability of different cleavable bispecifics.
Over the course of experiments we observed greater variations in activity for the BI-S and invBI-S SSOs, whereas splice-switching efficacy was relatively consistent for the monospecific ONs and the non-cleavable BI-C18 (data not shown). We hypothesize that these variations and slight loss of activity could be a result of cleavage of the disulfide linker, following several freeze/thaw cycles of aliquots. To assess this we exposed AONs to DTT overnight and ran them on denaturing polyacrylamide gels to assess their resistance to cleavage. For the BI-S SSO a large proportion of the parent AON was reduced to its monospecific components (Fig. 6b). In contrast, BI-C18 appeared to have complete resistance to DTT-mediated cleavage, as might be anticipated. If we subjected the BI-S compound to several freeze/thaw cycles, gel analysis demonstrated that the compound was already cleaved, even in the absence of DTT treatment (data not shown). Thus, while BI-S appears potent, it does seem to be less stable. To address this potential stability issue we exploited another approach to generate an internal disulfide (BI-XS) that produces monospecific oligonucleotides without 5′ or 3′ thiol groups upon intracellular cleavage and would be expected to have reduced rate of cleavage (Fig. 6c). In contrast to the previously used bispecific SSOs that are comprised of two 25mer targeting sequences, the BI-XS was designed to contain a 20mer sequence targeting the Dmd pre-mRNA, the rational being that a shorter sequence would be easier to synthesize in larger scale. Dose-response experiments comparing the 25mer and the 20mer Dmd exon 23 targeting sequence did not show any significant differences between the two SSOs, and so was thought not to affect overall efficacy (data not shown). Before assessing the activity of BI-XS, we validated that the XS linker, with an internal disulfide, could be cleaved in a reductive environment (i.e., using DTT). As can be seen in Fig. 6d, the disulfide was nearly completely cleaved in the presence of DTT, resulting in the release of two nonmodified separate SSOs. We next conducted dose-response experiments comparing BI-XS with the previously tested SSOs. All SSOs promoted dose-dependent exon skipping of exon 23 from the Dmd transcript (Fig. 6e). Similarly, using the lower 50 nM SSO concentration, all versions mediated myostatin skipping (Fig. 6f, g). Encouragingly, the use of an internal disulfide linker only partially reduced Dmd exon 23 skipping activity compared to the BI-S bispecific and monospecific SSOs, and was equal, if not better than the branched C18 linker SSO. For Mstn skipping, efficacy of the BI-XS was also reduced but only marginally compared to other AON chemistries assessed.
Discussion
The use of SSOs to modulate splicing has opened a new avenue for potential treatment of genetic diseases including SMA and DMD. For DMD, current clinical trials have focused on targeted skipping of exon 51, which as a single SSO would directly address the largest number of patient mutations (approximately 13% of all DMD patients). Similarly, single SSO targeting of exons 45, 44, and 53 would directly benefit 8%, 6%, and 8% of DMD patients respectively. By targeting pre-mRNA splicing therefore, an exon skipping approach has the advantage that it can address many different genomic DMD mutations. However, some mutations require more than a single exon to be targeted such as when a premature termination codon lies within an out-of-frame exon or when more than one exon is required to be skipped to restore the reading frame around a deletion mutation. Additionally, some DMD exons are particularly difficult to remove from the mature DMD transcript, and in these situations a cocktail of two or three SSOs targeting the same exon may be preferable (Aartsma-Rus et al., 2006; Adams et al., 2007). For these reasons, a multiple SSO approach may be preferable or indeed necessary. Indeed, taking this concept further, attempts have been made to develop a “one cocktail fits all” approach to address the major deletion “hotspot” within the central region of the DMD gene where it has been estimated that removal of the in-frame region of exons 45–55 could address 30%–60% of all DMD patients (AARTSMA-RUS, 2012). Extensive efforts have been made to devise efficient means to induce multiexon skipping. To date, cocktails of SSOs targeting different exons have been used but the collective skipping levels of the entire locus remain very low (van Vliet et al., 2008). This is perhaps unsurprising given that approach is made more difficult by the necessity not only for each individual AON to reach single nuclei, but also for the cocktail of AONs to bind simultaneously to the pre-mRNA prior to splicing reaction. Recently, Akoi et al. demonstrated for the first time relatively efficient multiexon skipping in the mdx52 mouse model using a cocktail of PMOs conjugated to a branched delivery vector composed of cationic guanidinium groups (Aoki et al., 2012). Although promising, toxicity concerns related to these types of delivery modalities remain a major obstacle.
In addition to the desire to simultaneously splice multiple exons from the DMD transcript, there is increasing evidence that targeting other genes involved in modulating the disease phenotype could be beneficial. DMD is associated with severe muscle wasting, fibrosis, altered muscle regeneration and, importantly, muscle degeneration whereby the muscle stem cell pool becomes exhausted. Therefore, additional therapies have been proposed to overcome these issues. For example, signaling induced by myostatin via the activin receptor (AcvR2b) is known to negatively regulate muscle growth by targeting down-stream myogenic regulatory factors in order to suppress proliferation (Thomas et al., 2000; Langley et al., 2002; McCroskery et al., 2003). This suggests that simultaneously restoring dystrophin function and reducing myostatin signaling would be advantageous. A number of approaches have been utilized to modulate the activity of the myostatin pathway, including myostatin neutralizing antibodies (Wagner et al., 2008), endogenous myostatin antagonists [myostatin propeptide (Bogdanovich et al., 2005), follistatin (Nakatani et al., 2008) and soluble Acvr2b (the myostatin receptor; Lee et al., 2001)], RNAi (Magee et al., 2006) and transcriptional gene silencing (Roberts et al., 2012a). Indeed, Dumonceaux and colleagues demonstrated that combination of myostatin suppression via RNAi and dystrophin rescue using U7-expressing adeno-associated viruses improved muscle function in mdx mice (Dumonceaux et al., 2010). Similarly, increased muscle size was observed when PMOs conjugated to a cell-penetrating peptide were administered into mdx mice as cocktails (Malerba et al., 2012).
To address the need for targeting multiple exons within the DMD gene, and to help further explore the potential of targeting genes involved in disease modulation, we sought to develop AONs that could incorporate dual targeting sequences. Dual-targeting SSOs have been developed previously to induce large multi-exon skipping of exons 45–51 within the DMD gene through use of a bispecific SSO with a 10-bp uracil spacing linker, compared to a cocktail of monospecific SSOs (Aartsma-Rus et al., 2004). Whilst both approaches could induce skipping, efficacy was low and results inconsistent such that no firm conclusions could be made as to which approach was more effective. Bispecific AONs have also been developed for suppressing gene expression of unrelated genes to target cancer (Rubenstein et al., 2012), but again, these bispecific AONs were interlinked by non-cleavable nucleotide hairpins. From a therapeutic efficacy point of view, we hypothesize that it would be preferable to incorporate a cleavable disulfide linker that would be reduced inside cells, leaving the two antisense sequences to bind their cognate RNA sequence (as depicted in Fig. 1). Indeed, Chung and coworkers have demonstrated the advantage of using disulfide-linked multimeric siRNAs for gene silencing applications (Chung et al., 2011). However, such dimeric and multimeric siRNAs are generated by conjugation in solution, leading to multiple products and purification steps that reduce the yields.
Based on this, we here developed a new method to generate bispecific SSOs internally spaced either by a non-cleavable hydrocarbon linker (BI-C18) or cleavable disulfide-based linkers (BI-S and BI-XS). In contrast to existing literature using bispecific AONs and siRNAs, we established a synthesis scheme that allows for synthesis of such AONs on solid-phase (Fig. 3). As seen in Supplementary Figs. S1 and S2, the methodology described here allowed bispecific SSOs to be efficiently synthesized with high purity. To verify the proof-of-principle of successful synthesis of these AONs, we demonstrate that these bispecifics could induce exon skipping for both Dmd and Mstn pre-mRNA transcripts (Fig. 4). We would hypothesize that a cocktail of monospecific SSOs that could localize to their individual pre-mRNA targets would have greater efficacy than a non-cleavable linker and this was indeed the case. These results are in accordance with results from similar bispecific SSOs spaced by uracil nucleotide linkers (Aartsma-Rus, personal communication) and is likely due to the fact that each bispecific can only bind to either of the two target pre-mRNAs at a given time. In contrast to the carbon linker, when using the BI-S version that contains a cleavable linker, skipping levels were dramatically increased, with efficacy equivalent or marginally exceeding the levels observed for cocktails of monospecifics. Due to the generally low levels of Dmd and Mstn transcripts in vitro, nested PCR analysis was required to assess levels of exon skipping. As such, measurement of skipping efficiency can only be deemed semiquantitative and we recognize the limitation of this approach. Nevertheless, it is clear that under the same assay conditions these comparative results emphasize the importance of intracellular cleavage of the two antisense components. To confirm that these bispecific AONs are cleaved under reducing conditions, each bispecific AON was exposed to DTT overnight and release of monospecific products assessed by gel electrophoresis. Demonstration that the C18 linker was not degraded, whereas as the BI-S and BI-XS were, is further evidence to support that cleavage in a reducing environment (such as in the cytosol) contributes to the enhanced efficacy.
Although the emphasis of this study was to develop a method of synthesizing dual-targeting AONs, simultaneous targeting of dystrophin to bypass disease-causing mutations, and myostatin to promote muscle hypertrophy, is a logical approach to address DMD. To this end, we sought to demonstrate a proof-of-principle and assess potency of bispecific versus monospecific AONs in vivo. At 7 days post intramuscular injection into the mdx mouse model, we observed considerable reduction in the levels of Mstn transcript containing exon 2 by RT-qPCR analysis in SSO treated muscles compared to untreated controls. As myostatin is a circulating signaling factor, we would not anticipate that intramuscular injection into a localized area would have a functional effect; however, this does provide proof-of-principle that a bispecific AON can have some function in vivo. Compared to the cocktail of individual SSOs, Mstn skipping activity was similar for the BI-S SSO indicating that at least for this particular gene target, that efficacy is not reduced when applying this strategy. Despite this success in targeting Mstn, we were unable to detect any exon 23 skipping of the Dmd pre-mRNA under any of the treatment conditions. As skipping could be observed for Mstn targeting we would anticipate that both the individual SSO and bispecific SSO would be taken up into the nuclei of muscle fibers. We can only hypothesize that the lack of skipping in vivo for exon 23 is due to insufficient nuclear concentration of AON to induce sufficient Dmd skipping to be detected by RT-PCR analysis. Additionally, we cannot rule out that difference between the Mstn and Dmd target genes, such as transcript levels and rates of turnover could attribute to the change in pattern of efficacy in vivo. Efficient delivery of charged nucleic acids to muscle is an unsolved challenge in the AON therapeutic field and it is likely that an effective delivery vehicles or high dosing would be required to perform a robust assessment of bispecific AONs in vivo. Nevertheless, for targeting Mstn, these results would suggest that a cleavable bispecific SSO has similar activity to separate SSOs following local delivery.
To address the question of how AON length may affect cellular uptake, we used Cy5-labeled SSOs to assess uptake into H2k mdx myotubes. Qualitative assessment by fluorescent microscopy suggested that bispecific and monospecific AONs were taken up by cells at similar levels (data not shown). However, when FACS analysis was performed, Cy5 signal was higher for cells transfected with monospecific SSOs (Fig. 5a). The reduction in Cy5 signal observed for the BI-S can be partly attributable to the reduced efficiency of the Cy5 coupling reaction to the longer AON, which we would estimate is about 70% as efficient (data not shown). A limitation of this approach is that we cannot distinguish between internalized and surface bound Cy5 label, however following washing steps we would expect the majority of the signal to be intracellular Cy5 signal. Importantly though, these results are highly suggestive that enhanced uptake of cleavable bispecifics is not the underlying reason of equivalent efficacy to monospecific SSOs. Given the novelty of using BI-S and BI-XS SSOs for a splice-switching approach, we assessed whether cell toxicity could be an issue. However, no notable cellular toxicity was present in any of the AONs used in this study for the concentration interval used for the in vitro experiments.
One obstacle we encountered over the course of experiments with the BI-S SSO was a partial loss in activity over time (data not shown). Therefore, we exploited an alternative linker dubbed XS (DTPA), originally developed for an alternative purpose (i.e., attaching oligonucleotide derivatives to gold particles). To our knowledge, the XS (DTPA) linker has not been used for this purpose before. The advantage compared to standard disulfide linkers is that it should be more stable outside the cellular environment and under reductive conditions be cleaved so as to not leave free thiol groups and therefore be of a more similar chemical nature as the monospecific SSOs (Fig. 6c). The BI-XS SSO dose-dependently induced exon skipping exceeding the activity of the non-cleavable bispecific (BI-C18). However, BI-XS displayed somewhat lower activity than monospecific versions or the parent BI-S SSO. This is most likely a consequence of the inability to fully cleave this SSO under reductive conditions. Indeed, following overnight exposure to DTT, the BI-XS AON appears more resistant to cleavage than the BI-S AON. Future studies with the BI-XS AON will focus on assessing its activity in vivo to determine if the apparent enhanced stability of the compound outweighs the observed reduction in exon skipping efficacy.
In conclusion, we have developed a robust system for solid phase synthesis of bispecific SSOs harboring internal disulfide interlinkages that are cleaved in reducing environment. We show that these are as least as potent as their monospecific counterparts. For more quantitative assessment of bispecific AON activity in vivo it is likely that a significantly higher dosing amounts over a longer time period would be necessary to induce a measurable functional improvement and these experiments along with in vivo testing of the BI-XS AON will be the subject of future studies. Additionally, it may also be prudent to measure the activity of bispecific AONs where the efficacy of the monospecific elements are more closely matched, or where the target transcripts have similar rates of turnover, as this would allow direct comparison of activity over a wider range of concentrations. Nevertheless, we clearly show that these bispecific oligonucleotides have robust in vitro activity and could prove very useful for multiexon skipping approaches, or multitargeting in general, where equal delivery of each antisense sequence is preferable. Also, from a regulatory therapy perspective, it is advantageous to have single therapeutic compounds over cocktails. Furthermore, we have recently been able to include further target sequences through generation of trispecific AONs, additionally comprising a miRNA-31 target site, since it has been implicated in regulation of dystrophin expression (Cacchiarelli et al., 2011), which will be further evaluated. Whilst the prospect for these bispecific or trispecific AONs is exciting, application in a preclinical or clinical setting will require the development of delivery vectors that allow for systemic delivery of these relatively large compounds and remains the greatest challenge for this approach.
Footnotes
Acknowledgments
S.ELA is supported by the Swedish Research Council (Vetenskapsrådet-med Unga Forskare) and the Swedish Society of Medical Research. CIES is supported by the Swedish Research Council. Waters (Sweden and United States) is greatly acknowledged for support and collaboration in the LC-MS area. We thank Tolga Sütlü at the Center of Hematology and Regenerative Medicine (HERM), Karolinska Institutet, for his kind help with the FACS analysis.
Author Disclosure Statement
The authors declare that there is no competing financial interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.