
Abstract
Plasminogen activator inhibitor-1 (PAI-1; SERPINE1) inhibits the plasminogen activators: tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). Elevated levels of PAI-1 have been correlated with an increased risk for cardiovascular disease. Pharmacologically suppressing PAI-1 might prevent, or successfully treat PAI-1 related vascular diseases. This can potentially be accomplished by using small RNA molecules (aptamers). This study's goal is to develop RNA aptamers to a region of PAI-1 that will prevent the ability of PAI-1 to interact with the plasminogen activators. The aptamers were generated through a systematic evolution of ligands via exponential enrichment approach that ensures the creation of RNA molecules that bind to our target protein, PAI-1. In vitro assays were used to determine the effect of these aptamers on PAI-1's inhibitory activity. Three aptamers that bind to PAI-1 with affinities in the nanomolar range were isolated. The aptamer clones R10-4 and R10-2 inhibited PAI-1's antiproteolytic activity against tPA and disrupted PAI-1's ability to form a stable covalent complex with tPA. Increasing aptamer concentrations correlated positively with an increase in cleaved PAI-1. To the best of our knowledge, this is the first report of RNA molecules that inhibit the antiproteolytic activity of PAI-1.
Introduction
P
To this end, numerous small molecule PAI-1 inhibitors have been identified (BROWN, 2010; FORTENBERRY, 2013). PAI-039 (Tiplaxtinin) is a well-characterized small molecule PAI-1 antagonist (Hennan et al., 2005; Hennan et al., 2008). It has been shown to decrease thrombosis formation and enhance the resolution of thrombus in vivo (Elokdah et al., 2004). Other PAI-1 inhibitors, such as TM5007 and TM5001, are also able to inhibit thrombus formation in vivo in both rats and non-human primates (Izuhara et al., 2008; Izuhara et al., 2010).
PAI-1 has three major functional domains: (1) the reactive center loop (RCL) region, (2) the vitronectin binding domain, and (3) the low-density lipoprotein receptor related protein site. Some of the currently available PAI-1 antagonists target more than one of PAI-1's domains. To understand the importance of each domain interaction, one must investigate them separately. Hence, we have designed small RNA molecules (aptamers) to independently target inhibitors to the various regions of PAI-1.
Aptamers are single-stranded nucleic acids, either DNA or RNA, that bind to their target protein with high affinity and specificity. Recently, we and others have developed PAI-1 specific aptamer inhibitors that disrupt PAI-1 from interacting with vitronectin (Blake et al., 2009; Madsen et al., 2010). Interestingly, neither of these aptamers inhibited PAI-1's antiproteolytic activity. In this study, we generated PAI-1 specific RNA aptamers that successfully disrupt the interaction of PAI-1 with tPA and can potentially be used as new PAI-1 antagonists.
Materials and Methods
Reagents
Human wild-type PAI-1 (wt PAI-1), which was produced in Escherichia coli, was purchased from Molecular Innovations, Inc. The human stable PAI-1 mutant containing the mutations K154T, Q319L, M354I, and N150H was also purchased from Molecular Innovations, Inc. These mutations “lock” PAI-1 in an active state and delay the spontaneous conversion of PAI-1 to its latent conformation. Our aptamers bind equally well to both wild-type PAI-1 and the stable PAI-1 mutant (see Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat). Consequently, the two molecules were used interchangeably in the studies and are referred to as PAI-1 throughout the manuscript. Human glycosylated PAI-1, produced in insect cells, was purchased from Molecular Innovations, Inc. The PAI-1/tPA complex protein was also purchased from Molecular Innovations, Inc., as was the human elastase cleaved PAI-1. Elastase cleaved PAI-1 is cleaved at the P3–P4 residues via immobilized elastase. This results in a nonreactive, reactive center loop inserted PAI-1 species. Elastase cleaved PAI-1 does not bind to plasminogen activator or to vitronectin. Recombinant PAI-1 vitronectin mutant containing a double mutation (Q123K and R101A) was generously supplied by Dr. Dan Lawrence (University of Michigan Medical School). This mutant molecule exhibit reduced binding to vitronectin. We purchased human monomeric vitronectin from BD Biosciences; Sperctrozyme tPA from American Diagnostica; and plasminogen activators, tPA, and uPA from Molecular Innovations.
Systematic evolution of ligands via exponential enrichment
As described previously, we developed RNA aptamers (Blake et al., 2009) using the systematic evolution of ligands via exponential enrichment (SELEX) method. The sequence of the starting RNA library (denoted Sel2 Library) was 5′-GGGAGGACGATGGG-N40-CAGACGACTCGCTGAGG ATCC-3′, where “N40” denotes random region 40-nucleotides in length. The RNA consisted of 2′-fluropyrimidines (2′-flurocytidine triphosphate and 2′-flurouridine triphosphate) rendering the RNA nuclease resistant. The initial library (R0) consists of approximately 1014 different RNA sequences. To develop aptamers to the site on PAI-1 that interferes with its interaction with plasminogen activators, we employed a toggling selection strategy involving PAI-1 and PAI-1/tPA complex. PAI-1 was the target protein in the initial round; however, in subsequent rounds we toggled between PAI-1 and PAI-1/tPA complex proteins. When the PAI-1/tPA complex was used as the target, the RNA molecules that did not bind to the protein were selected and the rest were discarded to eliminate the RNA molecules that bind outside of the RCL region. The RNA library was incubated with its respective protein for 15 minutes at 37°C, and we then separated unbound RNA from RNA/PAI-1 complexes by passing the mixture over a nitrocellulose membrane. The selection rounds 1–5 were performed in 3-[(3-cholamidopropyl)-1-propanesulfonate] (CHAPS) binding buffer E (20 mM Hepes, pH 7.4, 50 mM NaCl, 2 mM CaCl2, and 0.05% CHAPS), and rounds 6–10 were performed in CHAPS binding buffer F (20 mM Hepes, pH 7.4, 150 mM NaCl, 2 mM CaCl2, and 0.05% CHAPS). The RNA was eluted from the nitrocellulose by phenol/chloroform/isoamyl alcohol extraction followed by ethanol precipitation. In rounds with PAI-1/tPA, the eluent containing the unbound RNA was collected, the excessive buffer was removed, and the RNA was concentrated by butanol extraction. In both cases, the RNA was then subjected to reverse-transcription polymerase chain reaction. Briefly, approximately a quarter of the extracted RNA was reverse transcribed. The RNA was incubated with the 3′ primer in RT buffer for 5 minutes at 65°C and then cooled to room temperature. Afterwards, the dNTPs and AMV RT enzyme (Boehringer Mannheim) were added to the reaction followed by an additional incubation at 37°C for 45 minutes. The RT enzyme was deactivated by incubating the reaction at 95°C for 5 minutes. The RT product was then subjected to PCR, and the resulting complementary DNA library was concentrated and transcribed to RNA as described below. As we increased the selection rounds, the ratio of RNA to protein also increased. The initial ratio was 1:3 (protein:RNA). We increased the ratio with each round with the ratio being 1:10 in round 10.
In vitro transcription
The complementary DNAs were transcribed to RNA using a Dura Scribe T7 transcription kit (Epicenter Biotechnologies). Briefly, 2 μg of linearized template DNA and the T7 promoter were incubated with 100 mM dithiothreitol, 50 mM ATP, GTP, 2′-F-dCTP, and 2′F-dUTP in the presence of 10 mM Durascribe T7 enzyme mix. The reaction was then incubated at 42°C for 6 hours (or overnight) prior to adding deoxyribonuclease 1 (1 MBU) in order to remove the DNA template. We then extracted the transcript with phenol/chloroform/isoamyl alcohol. To check the transcript's purity, an equal volume of 2×formamide loading buffer was added and incubated at 65°C for 5 minutes. The RNA transcript was subsequently cooled to room temperature and subjected to electrophoresis on a 12% 7M urea denaturing gel. If the RNA transcript was pure and did not contain any smaller contaminating bands, we concentrated the RNA transcript using an oligonucleotide concentrator kit (Zymo Research). If not, we visualized the RNA by ultraviolet shadowing, excised the RNA band from the gel, minced, and incubated it in 2 mL Tris-EDTA buffer overnight at 4°C. The next day, we removed the RNA and concentrated it using Amicon Ultra centrifugal filters (Millipore). We determined the RNA concentration and then used it in subsequent experiments. Our RNA aptamers were incubated at 65–75°C for 5 minutes, followed by cooling (on ice) for 5 minutes before being used in all experiments.
Activity assays
All activity assays were performed in 96-well bovine serum albumin (BSA)–coated microtiter plates. Aptamer clones were heated at 65°C for 5 minutes and then incubated with PAI-1 (40 nM), or glycosylated PAI-1 (40 nM) in HNPN buffer (20 mM Hepes, 150 mM NaCl, 0.01%PEG, 0.0055% sodium azide) containing 2.5 mM CaCl2 at room temperature for 10 minutes. After this incubation, tPA (5–10 nM) or uPA (5–10 nM) was added and the reaction was then incubated for an additional 5–10 minutes at 37°C. Residual tPA activity was determined by cleavage of the chromogenic substrate, SPECTROZYME tPA (American Diagnostics) or S-2444 for uPA from diaPharma at a final concentration of 500 μM. After adding the substrate, the plate was immediately placed into a microplate reader (Versa Max). A kinetic reading was measured every 15 seconds for 20 minutes at an absorbance of 405 nm. Both tPA and uPA activities in the absence of the inhibitor were set at 100%.
In vitro complex formation
The aptamer clones (50–500 nM) were incubated with PAI-1 (500 nM) for 10 minutes at room temperature. Subsequently, tPA or uPA (400 nM) was added to the reaction and then incubated at 37°C for 5 minutes. The reaction was stopped by adding Laemmli sample buffer containing β-mercaptoethanol and boiled for 5 minutes. The samples were then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis, followed by Western blotting using either monoclonal PAI-1 antibody or polyclonal plasminogen activator (tPA or uPA) antibodies, or both.
Solid phase vitronectin studies
Solid phase vitronectin studies were performed as previously described (Blake et al., 2009). Briefly, 96-well microtiter plates were coated with 150 μL of vitronectin (Molecular Innovations, Inc.) at 250 ng/mL and incubated overnight at 4°C. The next day, the wells were washed three times with PBS-Tween (phosphate buffered saline with 0.01% Tween 20) and then dried at room temperature for 15 minutes. The plates were then blocked with 3% BSA for 1 hour at room temperature followed by washing three times with PBS-Tween. The plates were stored at 4°C until needed for the assays. For all assays, the aptamers were heated at 65°C for 5 minutes followed by cooling. PAI-1 (200 nM) was added to 96-well microtiter plates coated with vitronectin and incubated at room temperature for 30 minutes. The plate was washed three times with PBS-Tween, and then the aptamers (200 nM) were added to the respective wells and incubated for an additional 30 minutes. Next, 50 μL of tPA (2 nM) was added to each well and incubated for 30 minutes at room temperature. To determine residual tPA activity, 50 μL of SPECTROZYME tPA (500 μM) was added to each well and the plate was immediately placed into a microplate reader (VersaMax) set to read kinetically at 405 nm for 10 minutes (with readings every 20 seconds).
Binding assay and dissociation constant determination
The nitrocellulose double filter binding assay was used to determine binding affinities as previously described (Rusconi et al., 2000). Briefly, RNA was dephosphorylated using bacterial alkaline phosphatase (Gibco BRL), and the 5′ end was labeled with [γ-32P] ATP (PerkinElmer Life and Analytical Sciences, Inc.) using T4 polynucleotide kinase (New England Biolabs,). Target protein (PAI-1, elastase cleaved PAI-1, or PAI-1/tPA complex) at varying concentrations (0–5μM) was incubated with 5 μL of labeled RNA (2000 cpm/uL=0.1–0.2 nM) in binding buffer (20 mM Hepes, pH 7.4, 150 mM NaCl, 2 mM CaCl2, and 0.05% CHAPS) for 15 minutes prior to being added to the nitrocellulose membrane. The membranes were washed in wash buffer (20 mM Hepes, pH 7.4. 150 mM NaCl, 2 mM CaCl2) and the RNA that bound to the protein was retained on the nitrocellulose membrane. Unbound RNA was captured onto the nylon membrane. Total hot RNA on both the nitrocellulose and nylon membranes was determined by counting radioactivity using a liquid scintillation counter (Beckman Coulter, Inc.). The data were analyzed and the Kd was determined by fitting the data by nonlinear least squares (GraphPad Prism, GraphPad Software, Inc.) as a function of total protein concentration. The binding curves were fitted to the equation: B=Bmax [P]/Kd+[P] where B is the fraction bound, Bmax is the fraction of RNA bound at saturation, [P] is the protein concentration, and Kd is the dissociation constant. The raw data were corrected for nonspecific background binding of radiolabeled RNA to the nitrocellulose filter (corrected fraction binding). The background binding varied for each individual experiment; however, on average the background binding was less than 0.2%.
Results
Selecting RNA aptamers to the reactive center loop region of human PAI-1
Given the nature of aptamers, we suspect that they tend to preferentially bind to basic hydrophobic protein regions, such as the heparin or vitronectin binding domains. As our work and other studies have shown that targeting aptamers to PAI-1 generates molecules that bind to the vitronectin/heparin binding sites (Blake et al., 2009; Madsen et al., 2010). Therefore, we devised a toggled SELEX selection approach to minimize the number of RNA molecules that bind to these sites on PAI-1. This strategy allowed us to uncover molecules that could decrease the interaction of PAI-1 with plasminogen activators. Our starting RNA library containing 2′fluoro-pyrimidine modified bases was selected against PAI-1. In subsequent rounds, we alternated between PAI-1 and PAI-1/tPA complex. The bound RNAs were taken in rounds selected against PAI-1 (rounds 1, 3, 5, 7, and 9), while the unbound RNAs were taken in rounds selected against PAI-1/tPA (rounds 2, 4, 6, 8, and 10). We hypothesized that aptamer molecules that bind to the PAI-1/tPA complex will not bind with high affinity to a site on PAI-1 that is important for its interaction with tPA. Using PAI-1 in alternating rounds ensured that the RNA molecules retain their ability to bind with high affinity to the native protein. In Fig. 1A, we show an increased binding of the RNA library with increased PAI-1 protein concentration. From the binding curves, we calculated the binding affinities of the library after rounds 0, 2, 5, and 10 (Fig. 1A) as described in the material and methods section. The binding affinity of the individual libraries to PAI-1 increased with progressive rounds, indicating that they are enriched with molecules that bind specificity to PAI-1 (Fig. 1A). We then cloned and sequenced the round 10 RNA library, and isolated individual RNA aptamer clones. Three dominant RNA sequences were identified—R10-2, R10-4, and R10-14 (Table 1). These RNA aptamer clones comprised approximately 90% of the total RNA library pool. Subsequently, we determined the binding affinity of these individual clones to PAI-1. Fig. 1B shows the binding curves for the clones to PAI-1. All three bound to PAI-1 with affinities in the nanomolar range (Table 1). The aptamer clone R10-4 bound with the highest affinity to PAI-1.
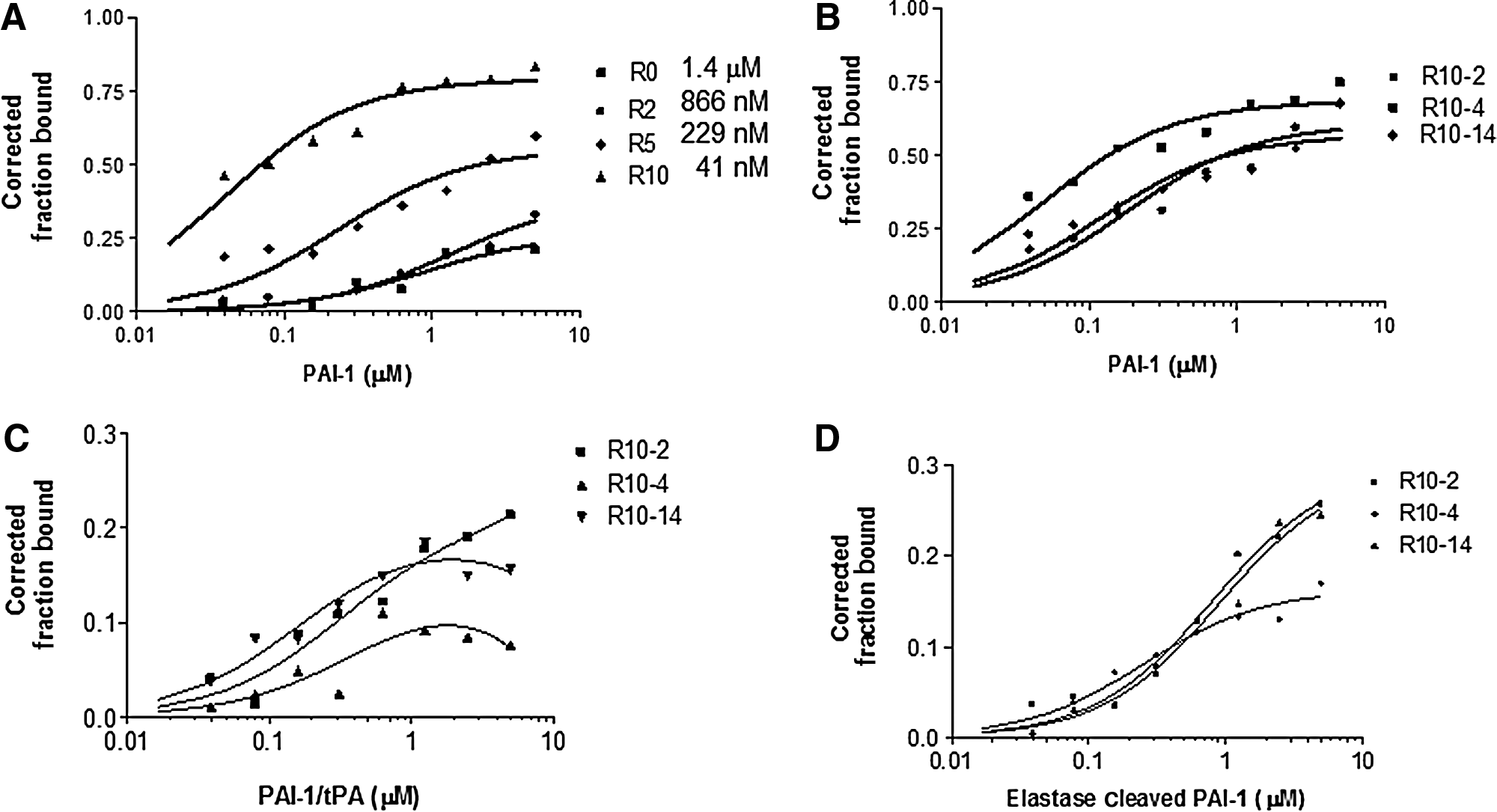
Toggle systematic evolution of ligands via exponential enrichment (SELEX) generated aptamers that bind to plasminogen activator inhibitor-1 with high affinity.
Frequency (percentage) in the round 10 (R10) library.
Kd, dissociation constant; PAI-1, plasminogen activator inhibitor-1 (non-glycosylated); EC-PAI-1, elastase cleaved PAI-1; gPAI-1, glycosylated PAI-1; PAI-1/tPA, PAI-1/tissue-type plasminogen activator complex.
To determine where on PAI-1 the aptamers are possibly binding, we assessed the binding of the aptamers to the PAI-1/tPA complex protein. We show in Fig. 1C that the binding of the aptamers to PAI-1/tPA was significantly decreased compared to the binding of the aptamers to PAI-1 (Fig. 1B). We saw a decrease in binding to all three aptamer clones (Fig. 1C; Table 1). These results suggest that our aptamers bind to a region of PAI-1 that is not accessible when in complex with tPA. This site could conceivably be the reactive center loop region. To evaluate this possibility further, we gauged their ability to bind to elastase cleaved PAI-1. Elastase cleaved PAI-1 is a species cleaved at the P3-P4 residues; thereby, rendering it nonreactive. Elastase cleaved PAI-1 does not bind to the plasminogen activators. The binding of the molecules to elastase cleaved PAI-1 would imply that the aptamers are interacting with a region on PAI-1 that is beyond the reactive center loop, and not in an area that is important for the binding of plasminogen activators. All three aptamers bound weakly to elastase cleaved PAI-1 (Fig. 1D; Table 1), further suggesting that they potentially bind to PAI-1 close to or in the vicinity of PAI-1's reactive center loop. However, it is possible that the decrease in the binding affinity of the aptamers to the elastase cleaved PAI-1 or the PAI-1/tPA complex compared with PAI-1is due to a conformational change in the protein rendering the aptamers unable to bind to “relaxed” PAI-1. Consequently, further structural studies are needed to determine the exact binding of the aptamers to PAI-1. Lastly, we determined if these molecules could bind to tPA alone. The binding of the clones to tPA was not substantial, thereby confirming that they bind explicitly to PAI-1 (not shown).
The aptamers do not compete with vitronectin for binding to PAI-1
Previously, we developed PAI-1 aptamers that bind to PAI-1's vitronectin/heparin binding site (Blake et al., 2009). To determine if these new PAI-1 aptamers also bind to this site, we assessed if vitronectin and the aptamers compete for binding to PAI-1 using a double binding nitrocellulose binding assay (see “Materials and Methods”). For this assay, a fixed concentration of PAI-1 was used, while varying the concentration of vitronectin. We display data for the R10-4 aptamer clone, since it binds to PAI-1 with the highest affinity. In Fig. 2A, we demonstrate how R10-4 retains its ability to bind to PAI-1 effectively, even at increasing concentrations of vitronectin. We then tested its ability to bind to the PAI-1/vitronectin mutant. The ability of vitronectin to bind to this PAI-1 mutant is reduced compared with PAI-1. Consequently, we rationalized that if R10-4 binds to the vitronectin binding site, we expect to see reduced binding of the aptamer to the PAI-1/vitronectin mutant. We show that R10-4 binds equally well to both PAI-1 and the PAI-1/vitronectin mutant (Fig. 2B). On the other hand, the binding of the PAI-1 aptamer SM20, which we have previous shown to bind to PAI-1's vitronectin/heparin binding site (Blake et al., 2009), exhibits reduced binding to the PAI-1/vitronectin mutant (Fig. 2B). The SM20 aptamer binds to PAI-1 with similar affinity as R10-4; however, these two molecules are binding to different PAI-1 domains (Fig. 2B). Similar results were found with both R10-2 and R10-14 (Supplementary Fig. S2). These data suggest that, unlike our previous aptamers, our new PAI-1 specific RNA aptamers bind to a site on PAI-1 that is outside of the vitronectin binding site.
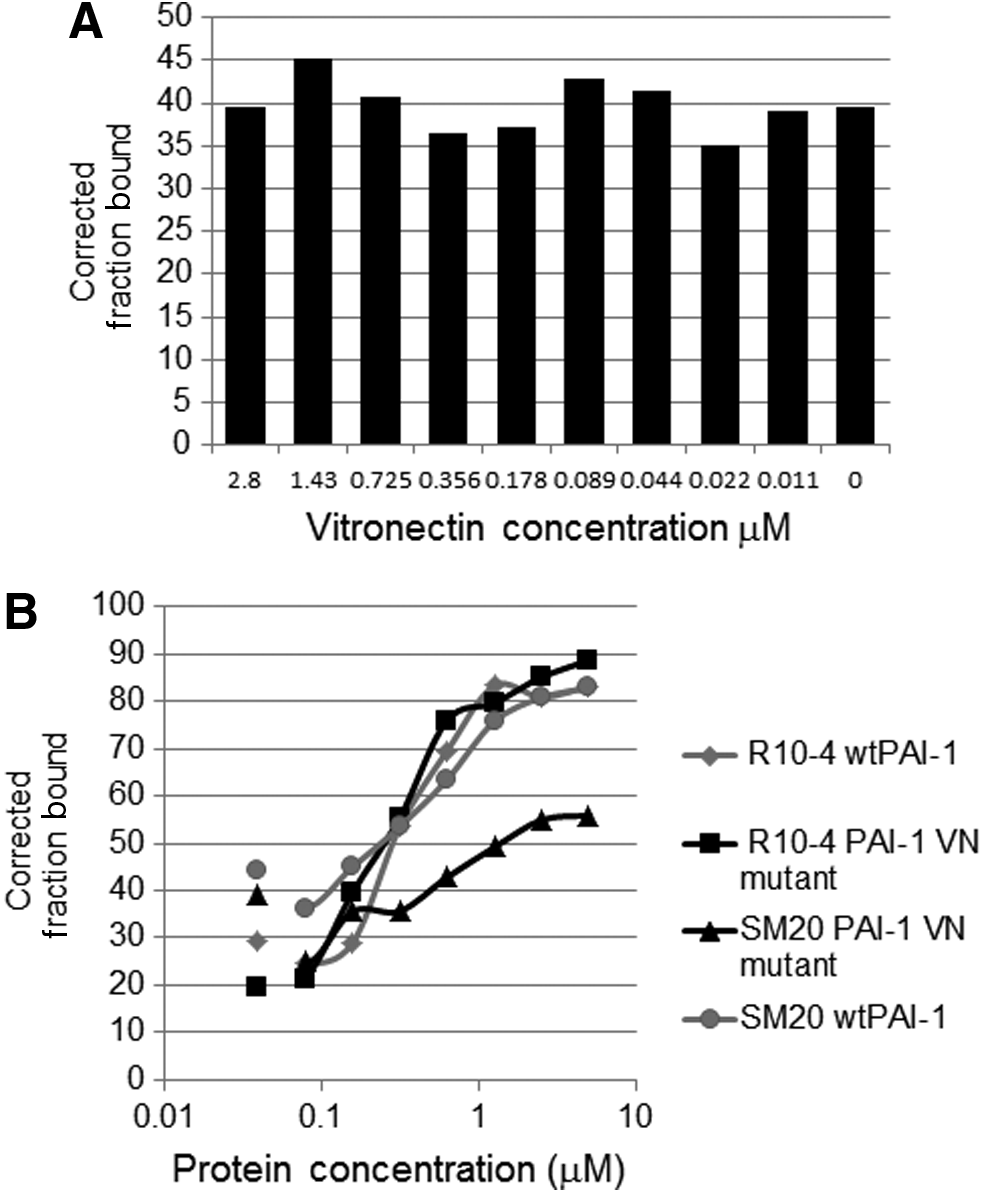
The binding of R10-4 to PAI-1 in the presence of vitronectin.
Aptamers attenuate PAI-1's ability to inhibit tPA activity
We next determined the ability of PAI-1 to inhibit the activity of tPA in the presence of the aptamers using a chromogenic assay. As expected, PAI-1 effectively inhibited tPA activity (Fig. 3A); however, in the presence of R10-4, PAI-1 is much less effective at inhibiting tPA (Fig. 3A, black bars). This inhibitory effect was observed at concentrations as low as 50 nM. The reaction at the concentrations tested appears to be saturated which is not surprising given that the concentration used is close to the Kd for R10-4 (Table 1). The aptamer clone R10-2 was less effective compared with R10-4 (Fig. 3A, grey bars). Neither aptamer completely inhibited PAI-1's activity. Interesting, the aptamer clone R10-14 exhibited no effect on PAI-1 activity even at a concentration of 2 μM (Fig. 3B). We used higher concentrations of aptamer in Fig. 3B, as we detected no effect of R10-14 at the lower concentrations. We then wanted to determine if the aptamers had a baseline effect on tPA activity in the absence of PAI-1. As shown in Fig. 3 for all three aptamers (aptamer alone lane; 100 nM), no significant aptamer related effect on tPA activity was detected (Fig. 3 A–B), again confirming that our aptamers bind specifically to PAI-1. In the above experiments, we pre-incubated PAI-1 with the aptamers, thereby allowing the aptamers to interact with PAI-1 in the absence of tPA. In order to eliminate this bias, we evaluated the effect of R10-4 on PAI-1 activity without being first incubated with PAI-1. To this end, we incubated PAI-1, R10-4, and tPA together for 10 minutes prior to adding the substrate. We show that R10-4 retains its ability to inhibit PAI-1 in a concentration dependent manner (Fig. 3C). However, the inhibition is slightly less effective compared to Fig. 3A.
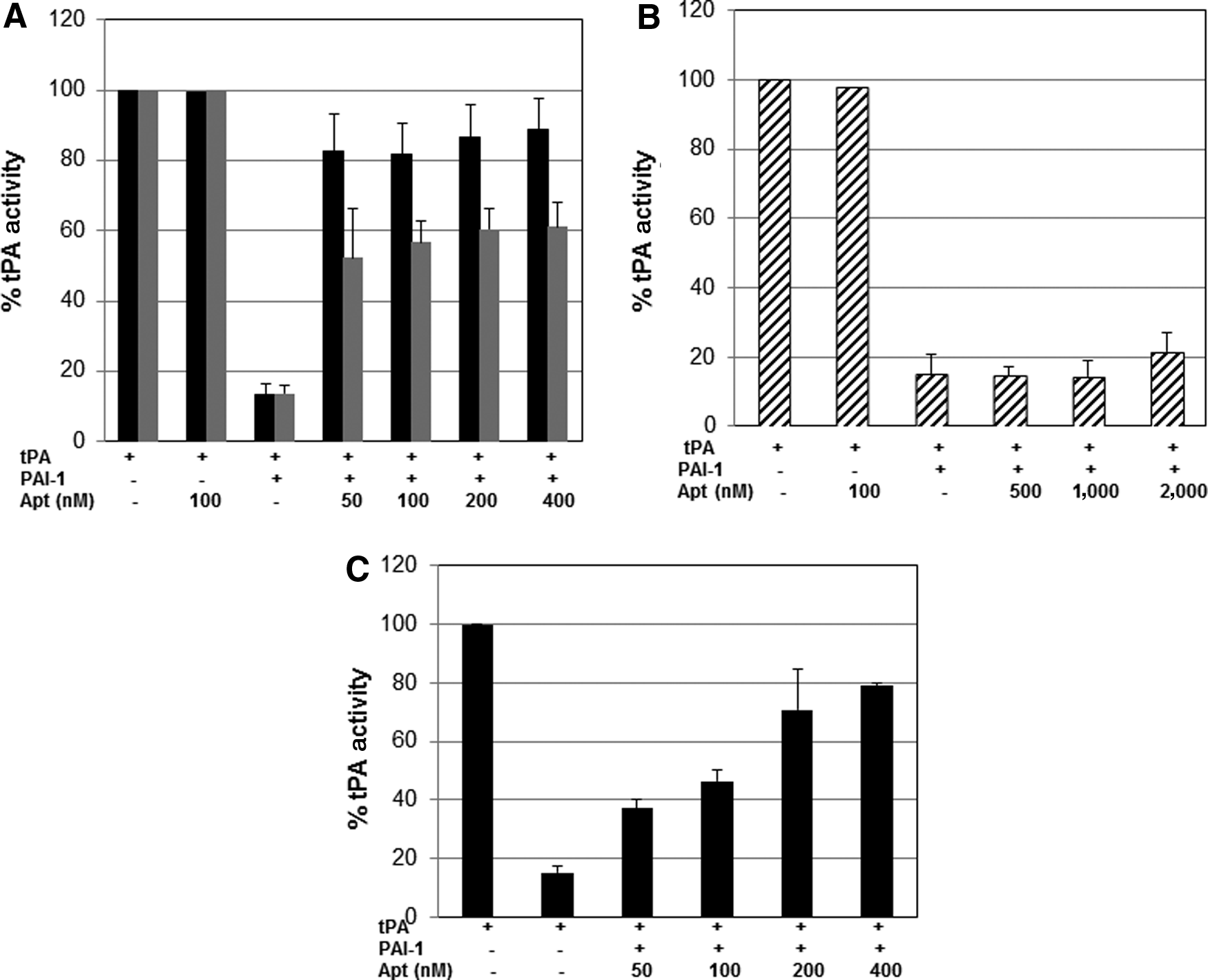
Effects of the RNA aptamers on PAI-1's ability to inhibit tPA.
We also assessed if the aptamers' ability to inhibit PAI-1 is diminished significantly over time. To this end, we determined the capacity of R10-4 and R10-2 to inhibit PAI-1activity at various time points. R10-14 was not tested since it did not appear to inhibit PAI-1's antiproteolytic activity against tPA. As expected, PAI-1 activity in the absence of the aptamers was sustained. Also, tPA remained completely active without PAI-1 (Fig. 4). In the presence of R10-4 and R10-2, PAI-1 inhibition of tPA was attenuated and we observed no significant change in the ability of these aptamers to inhibit PAI-1 activity during the timeframe of this experiment (Fig. 4). These data imply that the binding of the aptamers to PAI-1 is relatively stable. These data also indirectly suggest that tPA is unable to displace the aptamers from PAI-1. The ability of the aptamers to displace tPA from PAI-1 was not addressed; however, future studies will be aimed at better determining if the aptamers can regenerate tPA activity once it is in complex with PAI-1.

Effects of RNA aptamers on PAI-1's ability to inhibit tPA over time. The aptamer clones (R10-2 and R10-4) were heated at 65°C for 5 minutes and then incubated with PAI-1 (40 nM) for 10 minutes at room temperature. Afterwards, tPA (5 nM) was added and incubated at various times (0-60 minutes) at 37°C. Residual tPA activity was determined by cleavage of a tPA chromogenic substrate. The activity of tPA (asterisk) in the absence of PAI-1 was retained at 100% while in the presence of PAI-1; tPA activity was significantly inhibited (triangles). Incubating R10-4 with tPA alone did not significantly reduce tPA activity (diamonds). The effects of R10-4 (squares) and R10-2 (circles) on PAI-1's antiproteolytic activity were not considerably altered with time. Each data point was performed in triplicates and the experiments were repeated at least three times with similar results.
Aptamers inhibits glycosylated and non-glycosylated PAI-1
PAI-1 exists in either the glycosylated or non-glycosylated form. PAI-1 has three potential N-linked glycosylated sites (Asn209, Asn265, and Asn329). The inhibitory capacity of PAI-1 inhibitors can be glycosylated dependent, particularly with regards to monoclonal antibodies (Gils et al., 2003; Van De Craen et al., 2012). Consequently, it is essential to determine whether potential therapeutic molecules are able to inhibit both non-glycosylated and glycosylated PAI-1. We show that R10-4 and R10-2, but not R10-14, are able to inhibit glycosylated PAI-1 from suppressing tPA activity (Fig. 5). The glycosylated PAI-1 used in these experiments was produced in insect cells, and it is therefore possible that the glycosylated PAI-1 produced in human cells may differ. Nevertheless, based on previous studies, we do not expect this to be the case (Gils et al., 2003). Furthermore, all three aptamers bound to glycosylated PAI-1 at affinities that are comparable to non-glycosylated PAI-1 (Table 1).
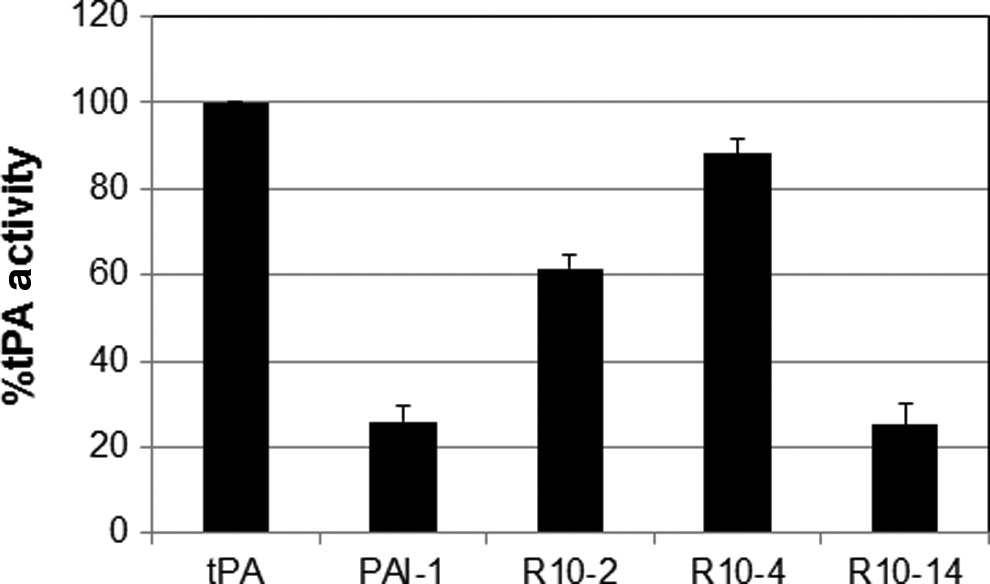
Effects of the RNA aptamers on glycosylated PAI-1. Glycosylated PAI-1 (40 nM) was incubated with the aptamer clones (200 nM) for 10 minutes at room temperature prior to adding tPA (5 nM). Residual tPA activity was determined by cleavage of a tPA chromogenic substrate. The activity of tPA in the absence of glycosylated PAI-1 was set to 100%. The data were performed in triplicates and the experiments were repeated at least three times. Error bars are standard deviation of the data.
Aptamers disrupt covalent complex formation and enhance the cleavage of PAI-1
PAI-1 forms a SDS-stable high molecular weight complex with tPA in the absence of inhibitors (Fig. 6A). Incubating PAI-1 with aptamer R10-4 either results in the loss of the high molecular weight complex or prevents the complex from forming (Fig. 6A). In either case, R10-4 prevents PAI-1 from interacting with tPA. Moreover, R10-4 increased the cleaved form of PAI-1 (Fig. 6A). Even in the absence of the aptamer we detected cleaved PAI-1. This could be due to the instability of PAI-1; still, a difference in the cleaved PAI-1 was apparent in lanes containing R10-4 (Fig. 6A). Given that we did not detect an immunoreactive band that corresponds to the complex between PAI-1 and tPA in the presence of the R10-4, we then determined whether we could identify a complex at lower concentrations of aptamer. At decreased aptamer concentration, we identified a high molecular weight complex band; however, the intensity of the bands is reduced compared with the no aptamer lane (Fig. 6B). Also, there is an increase in cleaved PAI-1 even at these lower concentrations, further confirming that the R10-4 promotes cleavage of PAI-1. We then wished to determine if a decrease in the PAI-1/tPA complex results in an increase in free tPA which would provide additional evidence that the loss of complex originate from disrupting the PAI-1/tPA interaction. To accomplish this, we probed the blot with an antibody to tPA. In the absence of aptamers, the band that corresponds to tPA was significantly diminished (Fig. 6C, lane 2); however, in the presence of R10-4, an increase in tPA is apparent (Fig. 6C, lanes 3 and 4). Figure 6 (lane 2) also shows a nonspecific band which is potentially a cleaved PAI-1 fragment.

Aptamer decreases the formation of the high molecular weight complex between PAI-1 and tPA.
To confirm that R10-14 does not have an effect on PAI-1's ability to inhibit tPA, we evaluated if R10-14 disrupts the PAI-1/tPA complex. R10-14 does not disrupt the PAI-1/tPA complex, which corroborates our activity assay data. Also, R10-14 does not enhance the cleavage of PAI-1 (Supplementary Fig. S3). Collectively, these data suggest that a potential mechanism by which R10-4 inhibits PAI-1 involves blocking its interaction with tPA and converting PAI-1 from a tPA inhibitor to a substrate for tPA.
The aptamer does not effectively inhibit PAI-1 bound to vitronectin
In plasma, active PAI-1 is unstable and converts spontaneously into its latent form. To slow down this transition, PAI-1 binds to vitronectin (Hekman and Loskutoff, 1985; Lindahl et al., 1989; Lawrence et al., 1997; Preissner et al., 1997). We wanted to determine whether the aptamer clones are able to inhibit PAI-1 effectively in complex with vitronectin. For these studies we used a solid phase vitronectin binding assay as described in the Materials and Methods section. PAI-1 was added to the coated plates and incubated for 30 minutes at room temperature. The PAI-1 solution was removed and the plates were washed in order to remove unbound protein. The aptamers were then added to the PAI-1/vitronectin complex, prior to adding tPA. In the absence of the aptamer, PAI-1 effectively inhibited tPA (Fig. 7). In contrast, R10-4 was less efficient at inhibiting PAI-1 when PAI-1 is in complex with vitronectin (compare with Fig. 3). Collectively, these results suggest that R10-4 is not as effective at inhibiting PAI-1 when bound to vitronectin.
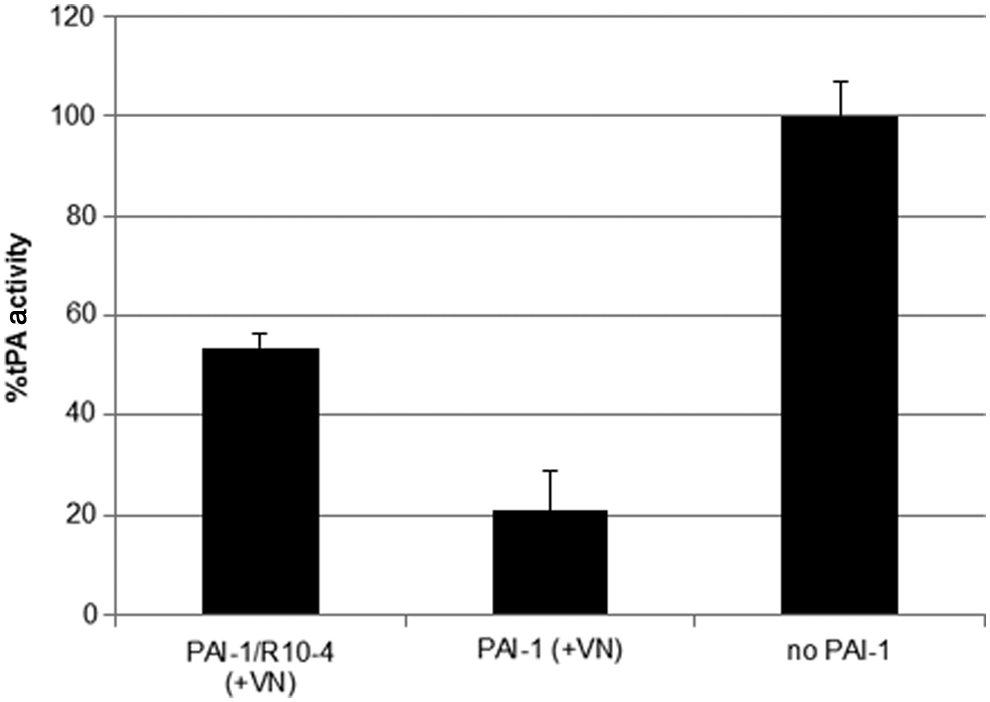
The capacity of R10-4 to inhibit PAI-1 is attenuated in the presence of vitronectin. PAI-1 (200 nM) was added to vitronectin coated plates and incubated at room temperature for 30 minutes. Afterwards the solution was discarded and the plates were washed three times with phosphate buffered saline–Tween. R10-4 (200 nM) or buffer was then added to the plate and incubated at room temperature for 30 minutes followed by adding tPA (2 nM) and incubating for an additional 30 minutes. Residual tPA activity was determined by cleavage of chromogenic substrate. The data on the y-axis is presented as percent tPA activity with tPA in the absence of PAI-1 being set at 100%.
Aptamers do not inhibit PAI-1's ability to suppress uPA activity
Considering that both tPA and uPA interact with PAI-1 in the same region, we then sought to determine the ability of these RNA aptamer molecules to alter PAI-1 inhibitory activity towards uPA. To our surprise, none of the aptamers inhibited PAI-1's antiproteolytic activity against uPA. Only a marginal affect at very high concentrations of the aptamer was observed (Fig. 8A). To further confirm these data, we show that the high molecular weight complex between PAI-1 and uPA is not disrupted in the presence of the aptamers (Fig. 8B). These results suggest that our aptamers bind to a site on PAI-1 that is important for its interaction with tPA, but not with uPA.
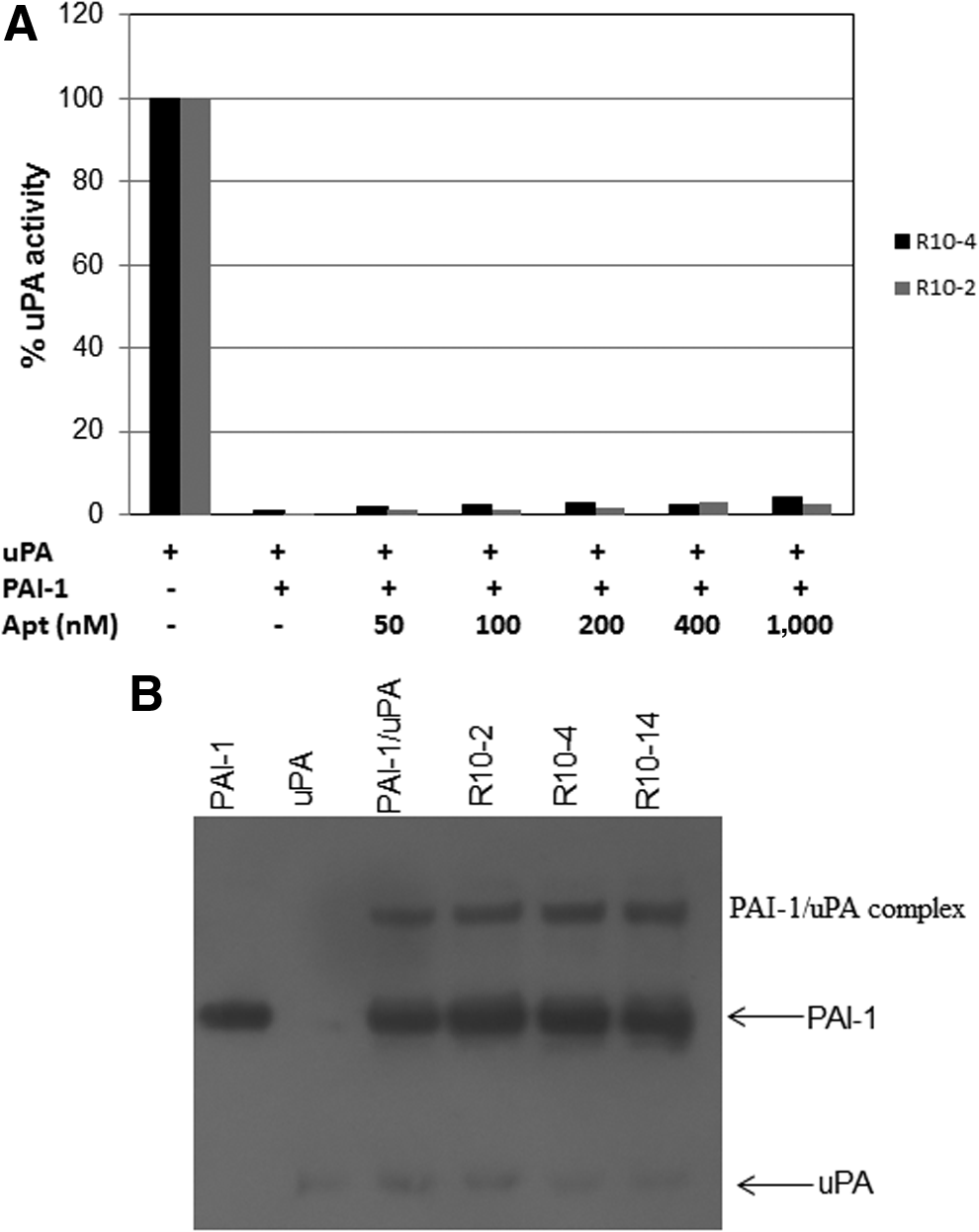
The aptamers do not attenuate PAI-1's ability to inhibit urokinase-type plasminogen activator (uPA).
Discussion
In this study, we have isolated and characterized a novel PAI-1 inhibitor that specifically disrupts PAI-1 from interacting with tPA. To our knowledge, this is the first report of an RNA oligonucleotide that disrupts the antiproteolytic activity of PAI-1 against tPA. We and others have previously described and discussed RNA aptamers that disrupt PAI-1 from interacting with vitronectin, but neither of these molecules was able to alter PAI-1 binding to tPA or uPA (Blake et al., 2009; Madsen et al., 2010). To accomplish this, we devised a selection protocol, using the PAI-1/tPA complex as the negative target to generate molecules that specifically bind near a site on PAI-1 that is important for its interaction with tPA. This site is potentially in the vicinity of the reactive center loop region. Adapting this bias strategy could potentially result in eliminating aptamers that compete with tPA for binding to PAI-1. However, given the nature of the aptamers, we rationalized that using this approach gave us the best chance of developing aptamers to this region. Results from both our binding and PAI-1 activity data imply that these molecules bind to the region on PAI-1 that is critical for inhibiting tPA. Notably, we have shown that these molecules do not disrupt the binding of PAI-1 to vitronectin. Consequently, similar to our first PAI-1 inhibitor (Blake et al., 2009), we have successfully developed RNA molecules that disrupt one function of PAI-1 without disrupting its other functions.
The link between cardiovascular disease and fibrinolysis is well established; elevated PAI-1 levels are known to be associated with an increased risk of cardiovascular diseases (Hamsten et al., 1985; Stefansson et al., 2003; Durand et al., 2004; De Taeye et al., 2005). This makes generating a PAI-1 antagonist very attractive. Several PAI-1 inhibitors have been developed and characterized for potential therapeutic usages (BROWN, 2010; FORTENBERRY, 2013). Numerous monoclonal antibodies have been shown to inactivate PAI-1, resulting in protection against thrombosis (Gils and Declerck, 2004); however, some are hampered by their immunological properties (van Giezen et al., 1997). Aptamers, have several advantages over antibodies: (1) aptamers are smaller and are able to interact with hidden protein epitopes; (2) aptamers can be chemically synthesized; (3) their stability and pharmacological properties can be modified; and (4) to date, they are non-immunogenic. In light of the specific challenges associated with antibodies, several researchers have focused on developing small molecule inhibitors (BROWN, 2010). One of the most characterized PAI-1 inhibitors is PAI-039 (tiplaxtinin).
Unlike our aptamers, PAI-039 inhibits PAI-1 from interacting with both tPA and uPA (Gorlatova et al., 2007). Other small molecule inhibitors, TM5275 and TM5007, also appear to preferentially target tPA (Izuhara et al., 2008; Izuhara et al., 2010; Yasui et al., 2013). The mechanism by which PAI-039 inhibits PAI-1 involves the inability of PAI-1 to form a stable complex with tPA/uPA, which correspondingly converts PAI-1 from a substrate to an inhibitor. Our aptamer molecules appear to inhibit PAI-1 via a similar mechanism, as discussed above. Similar to PAI-039, R10-4 and R10-2 are able to inhibit both non-glycosylated and glycosylated PAI-1. PAI-039 is unable to inhibit PAI-1 in complex with vitronectin (Gorlatova et al., 2007). One suggestion for this is that PAI-039 makes contact with the vitronectin binding site, thereby sterically blocking vitronectin from binding to PAI-1. This is not the case with the aptamers studied here. In Fig. 2, we show that vitronectin does not compete with R10-4 for binding to PAI-1, suggesting that the R10-4 and vitronectin bind to different PAI-1 domains. It is conceivable that R10-4 binds to the PAI-1/VN complex but with reduced affinity. We detected partial inhibition of PAI-1 in the presence of vitronectin, but clearly the ability of R10-4 to inhibit PAI-1 in complex with vitronectin is attenuated (Fig. 7). Nevertheless, unlike our previous PAI-1 aptamers (Blake et al., 2009), R10-4 does not bind directly to the PAI-1 vitronectin/heparin binding site. Therefore, we believe that these aptamers do not span to the vitronectin binding site. However, as with PAI-039, our aptamers' ability to inhibit the PAI-1/vitronectin complex is decreased.
PAI-1 inhibits its target protease by a substrate suicide mechanism. The protease recognizes the reactive center loop site of the inhibitor and erroneously assumes that it is a substrate. The protease then cleaves the P1–P1′ bond, creating an ester bond between the protease's active site Serine, and the P1 serpin residue. This results in an acyl enzyme intermediate. The N-terminal portion of the RCL is then inserted into the large central beta sheet A of the serpin, eventually becoming a new strand in the serpin's A beta-sheet (beta-strand s4A). The protease is then “dragged” to the opposite pole of the serpin, and during this transition, the active site of the protease is distorted and rendered inactive. When the RCL insertion is slowed, thereby compromising the ester bond hydrolysis, the protease is released and the serpin is cleaved. In this situation, the serpin then behaves like a substrate. Our results show that in the presence of R10-4, PAI-1 is cleaved, suggesting that it converts PAI-1 from an inhibitor of tPA to a substrate for tPA. Several RNA aptamers bind to sites and sterically hinder proteins from interacting with their targets (Jeter et al., 2004; Long et al., 2008). The thrombin RNA aptamer interferes with the ability of thrombin to interact with macromolecules, such as heparin (Long et al., 2008; Bompiani et al., 2012). While we cannot pinpoint the exact binding location of our aptamers on PAI-1, our results lead us to propose that they bind to a site near the reactive center loop region. We further suspect that they interfere with the hydrolysis of the ester bond and the translocation of the protease to the opposite pole of the serpin. Consequently, we hypothesize that the aptamers are preventing the conformational change in the serpin that is necessary for RCL insertion and protease inhibition. Additional structural studies are required to determine definitively if this is indeed the mechanism.
The discovery that our aptamers are able to alter PAI-1's interaction with tPA, but not uPA, suggests that they make contact with an area on PAI-1 that is not directly involved in the interaction of PAI-1 with uPA. This is an interesting and perplexing situation, given what we know about PAI-1's interactions with tPA and uPA. The reason may rest with the exosite interactions between the surface exposed variable region 1 (VR1; 37-loop) of tPA and uPA. It has been shown that the 37-loop of both tPA and uPA are important in the initial Michaelis complex formation (Tachias and Madison, 1997; Ibarra et al., 2004). The 37-loop of tPA contains more positively charged residues than the 37-loop of uPA, suggesting a more extensive interaction between PAI-1 and tPA (Lin et al., 2011). Hence, the aptamers may bind to a site on PAI-1 that is important for associating with the 37-loop of tPA, but not with the 37-loop of uPA. Another more conceivable explanation emanates from the strategy we used in selecting the molecules. Our bias strategy selected molecules that bind to the site on PAI-1 that is occupied by tPA. This highlights the specificity of aptamers. We do acknowledge that the mechanism by which these aptamers inhibit PAI-1 is not clear and additional studies are required to get a better understanding of the mechanism. Nonetheless, whereas PAI-1 inhibits both uPA and tPA, tPA is more important in thrombosis and fibrinolysis. Having a PAI-1 inhibitor that specifically targets the PAI-1 interaction with tPA is an ideal therapeutic agent and will permit further investigations into the importance of the PAI-1/tPA interaction in various disease states.
We hypothesize that the abatement of PAI-1's antiproteolytic function will be a beneficial therapeutic option for the treatment and prevention of PAI-1 associated vascular events. To date, there are no PAI-1 inhibitors available for clinical use. The use of aptamers to combat PAI-1 levels has several advantages over the currently available inhibitor, among which is the ability to inhibit one function of PAI-1 without disrupting its other functions. The use of aptamers as therapeutics is becoming a rational treatment option as evident from the number of aptamers currently in clinical trials (Germer et al., 2013; Li et al., 2013). In summary, the present study documents for the first time that RNA aptamers are able to disrupt antiproteolytic activity of PAI-1. These aptamers have the potential to be used in treating individuals that are prone to PAI-1 related cardiovascular events. In vitro and in vivo studies on these aptamer are ongoing to assess their full potential as a PAI-1 antagonist therapeutic agent.
Footnotes
Acknowledgement
This study was supported by grant HL096407 awarded to YMF from the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.