
Abstract
Antisense synthetic oligonucleotides have been developed as potential gene-targeted therapeutics. We previously reported polymerase–endonuclease amplification reaction (PEAR) for amplification of natural and 5′-O-(1-thiotriphosphate) (S)-modified oligonucleotides. Here, we extended the PEAR technique for enzymatic preparation of 2′-deoxy-2′-fluoro-(2′-F) and 2′-F/S double-modified oligonucleotides. The result showed that KOD and Phusion DNA polymerase could synthesize oligonucleotides with one or two modified nucleotides, and KOD DNA polymerase is more suitable than Phusion DNA polymerase for PEAR amplification of 2′-F and 2′-F/S double modified oligonucleotides. The composition of PEAR products were analyzed by electrospray ionization liquid chromatography mass spectrometry (ESI/LC/MS) detection and showed that the sequence of the PEAR products are maintained at an extremely high accuracy (>99.9%), and after digestion the area percent of full-length modified oligonucleotides reaches 89.24%. PEAR is suitable for synthesis of modified oligonucleotides efficiently and with high purity.
Introduction
T
It has been reported that unmodified oligonucleotides are highly susceptible to endogenous nuclease degradation in the serum, and their half-life in mouse whole blood is only approximately 10 min [4]. Moreover, unmodified oligonucleotides have some serious side effects; for example, the growth inhibitory oligonucleotides compete with the human telomere sequence oligonucleotides for binding to a specific cellular protein [5]. In addition, GC-rich oligonucleotides are prone to have nonspecific effects, such as immunostimulating and complement activation activities [6–10]. Therefore, unmodified oligonucleotides may cause serious adverse effects when they are used directly and extensively in human.
Fortunately, antisense oligonucleotides modified with certain appropriate chemical groups, such as 5′-O-(1-thiotriphosphate) (S), 2′-O-methoxyphenyl (2′-MOE), 2′-methyl (2′-OME), 2′-deoxy-2′-fluoro-(2′-F), and locked nucleic acid (LNA), exhibited increased specificity and stability in vivo, significantly enhanced antisense effects, and reduced side effects as compared with nascent oligonucleotides [11]. In recent years, there have been numerous reports on using modified oligonucleotides for gene-targeted therapy, in which modifying the alpha phosphate and the DNA backbone at the 2′-position of the furanose ring are the most useful in enhancing the affinity and the stability, improving the medicinal properties without toxicity [11,12]. For example, Hutvagner [13] reported that 2′-methyl (OME)-modified AONs can act as irreversible, stoichiometric inhibitors of small RNA let-7, provided an efficient and straight forward way to block small RNA function in human HeLa cells and nematodes. Modified oligonucleotides not only bind with the target nucleic acid more efficiently [14], enhance resistance to endogenous nuclease, and reduce immunostimulating activity [15]; they also promote the delivery of them into the disease tissues, such as spinal cord, liver, muscle, bone marrow, lung, blood, and solid tumors [16,17].
All along, oligonucleotides used in clinic or fundamental biomedical studies are mostly derived from chemical DNA synthesis by using the standard phosphoramidite method [18]. However, chemical synthesis method has some shortcomings, such as errors, impurities, and pollution [19]. Although by increasing the quantity of synthesized oligonucleotides and improving the purification methods would decrease the error rate to some extent, the room for improvement is limited due to the inherent constraints of chemical synthesis [20]. On the other hand, in chemical synthesis, some reactant, such as trichloroacetic acid, is considered as a potential carcinogen [21].
To overcome these problems, enzymatic reaction might be an alternative or better solution. The average error rate of ordinary DNA polymerase, such as Taq DNA polymerase, is as low as 7.2×10−5 [22], which is much lower than that of chemical synthesis method whose error rate is as high as 3×10−3 [20]. And the error rates of high fidelity DNA polymerases, such as Pfu, KOD, Phusion, and Vent DNA polymerase are even much lower [23]. In enzymatic reactions, most reactants including buffer solution, ions, and micromolecules are safe to human and environment, also they are easy to be eliminated.
In our previous studies, we developed a nucleic acid amplification technique, the polymerase-endonuclease amplification reaction (PEAR), and demonstrated that it is suitable for the enzymatic production of nature or modified antisense oligonucleotides with a high purity [24,25]. The two most popular modifications in gene silencing and anti-mRNA research are 2′-deoxy-2′-fluoro-(2′-F) and 5′-O-(1-thiotriphosphate)-(S). As the first-generation AONs, S-modification has been widely used in clinical studies, mostly because of their increased in vivo stability [26]. In addition, it was reported that 2′-F-modification increased the binding affinity of oligonucleotides against target RNA sequences [27], and significantly increased serum stability [28]. Partial 2′-F-RNA modification is active throughout the sense and antisense strands [29–31], and fully substituted nucleic acids induce RNA interference in mammalian cell culture [32]. In a previous study, we have prepared oligonucleotides with dATPαS, dGTPαS, or dCTPαS modifications by PEAR but found that dTTPαS could not be incorporated efficiently using Phusion DNA polymerase [25]. In order to prepare oligonucleotides with a wider range of modifications, here we tried other DNA polymerases.
As high-fidelity thermostable DNA polymerases, KOD DNA polymerase and Phusion DNA polymerase exhibit strong 3′→5′ exonuclease/proof-reading activity, an activity that lacks in Taq DNA polymerase. Moreover, both of them exhibit excellent processivity and elongation capability and extreme fidelity. The most important is that both of them can synthesize not only unmodified DNA but also modified DNA. Using KOD DNA polymerase, Kuwahara reported the synthesis of LNA-modified DNA [33]. Johannsen and others showed that 2′-amino-LNA (2′-amino-LNA-TTP) can be a good substrate for Phusion DNA polymerase [34].
In this study, we validated the PEAR technique for the preparation of 2′-F-modified and 2′-F/S double modified oligonucleotides using KOD DNA polymerase and Phusion DNA polymerase.
Materials and Methods
Materials
Four 2′-fluoro-2′-deoxyribinucleoside-5′-triphosphates (2′-F-dNTPs), including 2′-F-dATP, 2′-F-dCTP, 2′-F-dGTP, 2′-F-dUTP and four 2′-deoxyribonucleotides-5′-O-(1-thiotriphosphate) (dNTPαSs), including dATPαS, dGTPαS, dCTPαS, and dTTPαS, whose structural formula are shown in Fig. 1, were purchased from Trilink BioTechnologies, Inc. KOD DNA polymerase was purchased from TOYOBO (Shanghai) Biotech Co., Ltd. Phusion DNA polymerase, highly thermostable restriction enzyme PspGI, and dNTPs were purchased from New England Biolabs, Inc. UNIQ-10 Spin Column Oligo DNA Purification Kit was purchased from Sangon Biotech (Shanghai) Co., Ltd. Synthetic oligodeoxynucleotides, including a target (X) and a probe (P), were synthesized by Integrated DNA Technologies, Inc. and purified by high-performance liquid chromatography (HPLC). The sequence of X is 5′-TGT AAA CAT CCT CGA CTG GAA G-3′, which is derived from human microRNA hsa-miR-30a. The structure of P is X′R′X′R′X′, where X′ and R′ is complementary respectively to X and R. The sequence of P is 5′-CTT CCA GTC GAG GAT GTT TAC A

The diagram of the molecular structure of the substrates:
PEAR reactions
PEAR reactions were run in a 96-well Applied Biosystems 9700 Thermal Cycler, in a 100-μL volume reaction mixture containing 200 μM each of dNTP, 0.1 μM target, and 1.0 μM probe (or “seeds” PEAR products). For Phusion-based PEAR, 15 mM Tris-HCl (pH 8.0), 30 mM KCl, 5 mM (NH4)2SO4, 2.5 mM MgCl2, and 0.02% bovine serum albumin (BSA) were added; and for KOD-based reactions, the reaction mixture contains 120 mM Tris-HCl (pH 8.0), 1 mM MgSO4, 6 mM (NH4)2SO4, 10 mM KCl, and 0.1% Triton X-100, 0.01% BSA. In desired reactions, one or two natural dNTPs were completely replaced with the corresponding 2′-F-dNTPs or dNTPαSs. The concentration of DNA polymerase and PspGI for different kinds of modifications are shown in Table 1. For the second round of PEAR, the reaction conditions were the same as that of the first round, except that “seeds” (2 μL PEAR products) were added instead of the target and probe.
The PEAR reactions were initiated at 95°C for 1 min, followed by 35 cycles of denaturing at 95°C for 15 s, annealing at 55°C for 35 s, elongation, and cleaving at 75°C for 3 to 5 min. If desired, PspGI digestion of the products was conducted under 75°C for 1 to 16 h by adding 0.1 volume 10× NEBuffer 4, 0.4U/μL PspGI, and ddH2O to 2× volume. PEAR products were examined by nondenaturing polyacrylamide gel electrophoresis (PAGE) in 15% gels at 5 V/cm, stained with ethidium bromide, and detected by an ultraviolet illuminator. PEAR products were purified using UNIQ-10 Spin Column Oligo DNA Purification Kit according to its protocol to remove enzymes, BSA, and excessive dNTPs.
Mass spectrometry analysis of PspGI digested PEAR products
PEAR products were fully digested in a cleavage mixture containing 1× NEBuffer 4 and 1.0 U/μL of PspGI. Cleavage reactions were incubated for 8 h at 75°C. Before and/or after PspGI digestion, the products were purified as described above. Electrospray ionization liquid chromatography mass spectrometry (ESI/LC/MS) analysis was performed by Novatia, LLC using their high-throughput characterization system 35] to characterize the product oligonucleotides and profiling for components.
Results
PEAR amplification of 2′-F-dATP and 2′-F-dGTP modified oligonucleotides using Phusion DNA polymerase
We used one or two 2′-fluoro-modified dNTPs (2′-F-dNTPs) to substitute the corresponding nonmodified dNTPs in PEAR. The PAGE electrophoresis results are shown in Fig. 2A. Lane 1 is PEAR products amplified using unmodified dNTPs, wherein a series of DNA bands represent the tandem repeats of the target in different lengths. Lane 2 is amplified PEAR products with a modification of the 2′-F-dATP instead of the corresponding normal dATP. As in lane 1, a series of DNA bands that represent the different-length tandem repeats were found; however, when one of the four dNTPs, such as dATP (lane 6), is absent, the PEAR reaction is stopped completely. The yield of modified PEAR products depends on the number of cycles; the maximum yield of modified PEAR products is about 200 ng/μL, similar to the yield of nonmodified PEAR products. Using Phusion DNA polymerase, PEAR could incorporate 2′-F-dATP and 2′-F-dGTP into PEAR products. However, as shown in Fig. 2B, the exponential PEAR was almost abolished when dCTP and dTTP were replaced with 2′-F-dCTP and 2′-F-dUTP.
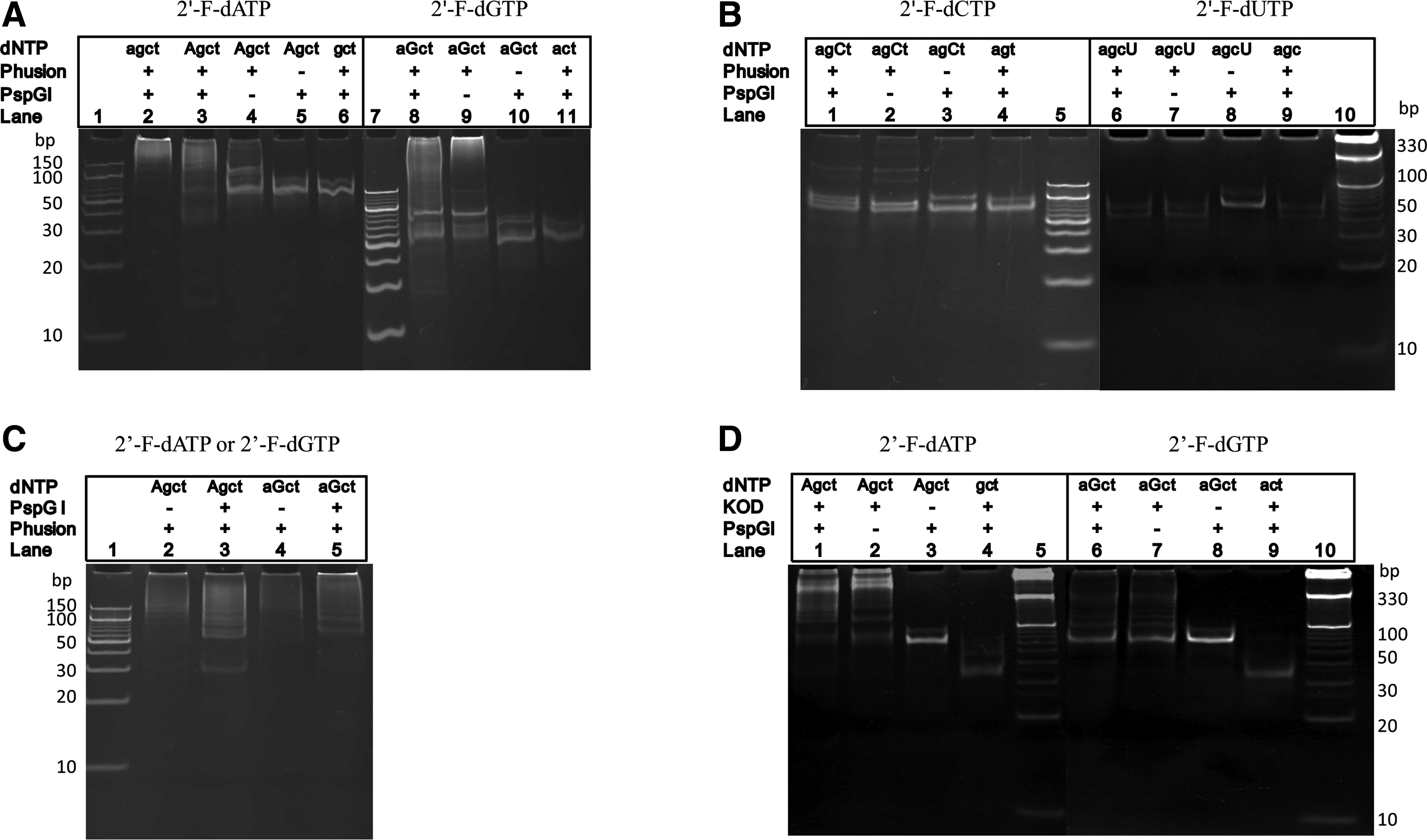
Polyacrylamide gel electrophoresis (PAGE) electrophoresis of the polymerase–endonuclease amplification reaction (PEAR) products. Lowercase letters (agct) represents unmodified dNTPs; uppercase letters (AGCT) represent modified dNTPs (2′-F-dNTPs or dNTPαSs).
PEAR amplification of 2′-F-dNTPs modified oligonucleotides using KOD DNA polymerase
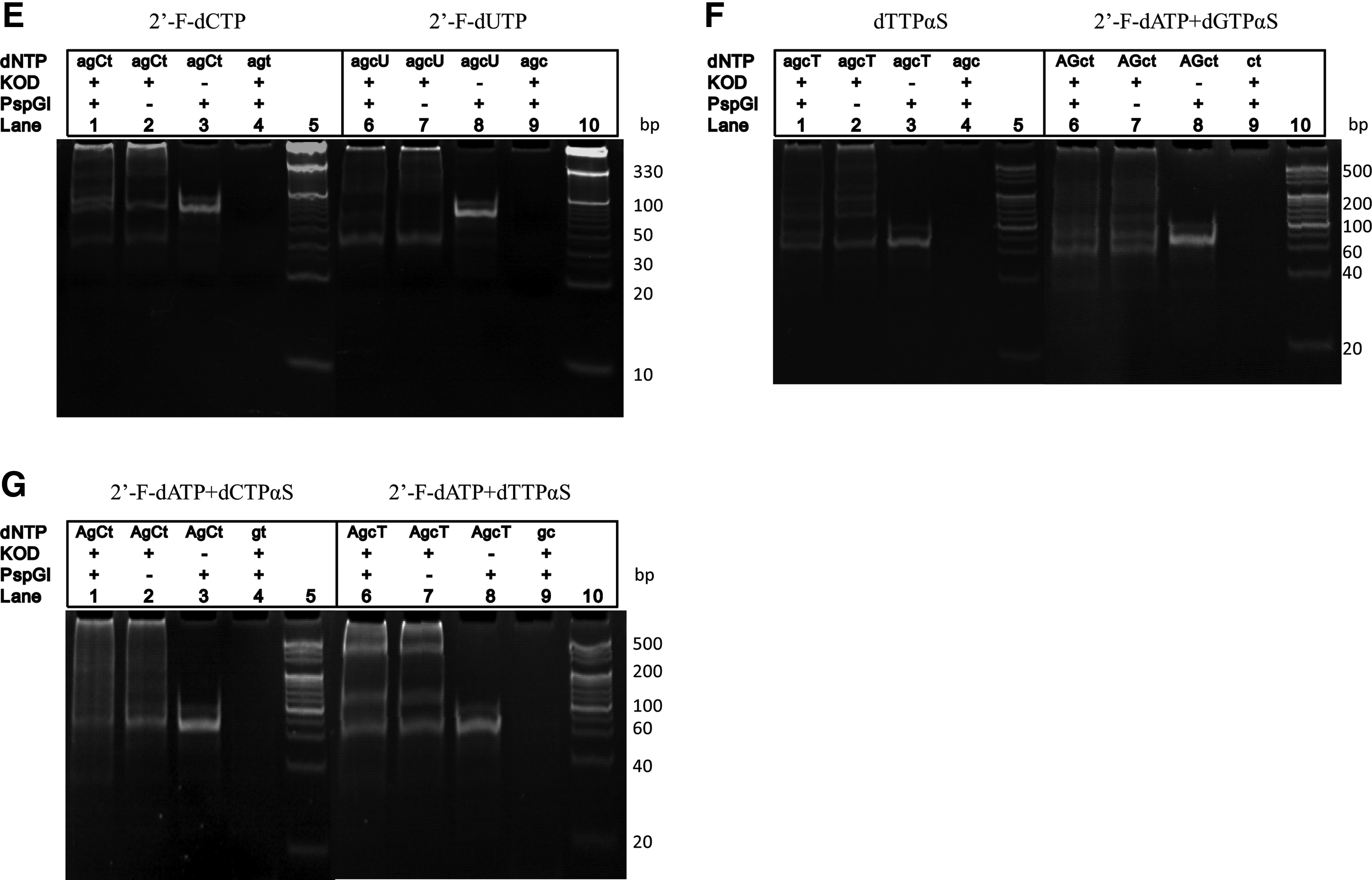
When KOD DNA polymerase was employed in PEAR instead of Phusion DNA polymerase with various kinds of 2′-F-modified dNTPs, the PEAR results showed that each of four 2′-F-dNTPs was successfully incorporated into PEAR products (Fig. 2D, E).
PEAR amplification of dTTPαS modified and 2′-F-dATP/dNTPαSs double-modified oligonucleotides using KOD DNA polymerase
In a previous study, we reported that dTTPαS could not be incorporated into desired oligonucleotides using Phusion DNA polymerase [25]. In the present study, using KOD DNA polymerase, dTTPαS was incorporated into PEAR products successfully, as shown in lane 1 and lane 2 in Fig. 2F. Since each of four dNTPαSs and four 2′-F-dNTPs were incorporated into PEAR products separately, we tried PEAR reactions with two different kinds of modification (such as dATPαS+2′-F-dGTP). As shown in lanes 6 and 7 in Fig. 2F and 2G, KOD DNA polymerase has the ability to synthesize double-modified oligonucleotides, including 2′-F-dATP+dGTPαS, 2′-F-dATP+dGTPαS, and 2′-F-dATP+dGTPαS.
Re-amplification by PEAR with the “seeds” of a previous round
As an advantage of PEAR, the products can be used as seeds for the next round of PEAR directly without any treatment. In such a reamplification reaction, an appropriate amount of PEAR products were added to substitute the target (X) and the probe (P) and keep the amount of other substances unchanged. As shown in Fig. 2C, lane 3, using 2′-F-dATP modified PEAR products as seeds for the PEAR reaction, lane 2 is a control with only Phusion DNA polymerase and without PspGI. Lanes 9 and 10 are the PEAR products amplified using 2′-F-dGTP modified PEAR products as seeds.
Characterization of the composition of PEAR products
We applied HP/LC/MS to analyze the relative molecular mass of the second-round PEAR products incorporated with the modification of 2′-F-dATP by KOD DNA polymerase and digested by PspGI. The determined molecular structure and component of the PEAR products were shown in Fig. 3 and Table 2. The PEAR products mainly consist of six strands, where the two unmodified chains are the original probe (probe B and probe C), and the other four modified oligonucleotides are the PEAR products. The components A, D, E, and F of PEAR products are shown in Fig. 3 and Table 2, whereas the other products are shown only in Table 2. Because the quantity of PspGI and the cleavage time are limited, some of the PEAR products (G and H) were not completely digested and thus still contained repeats.
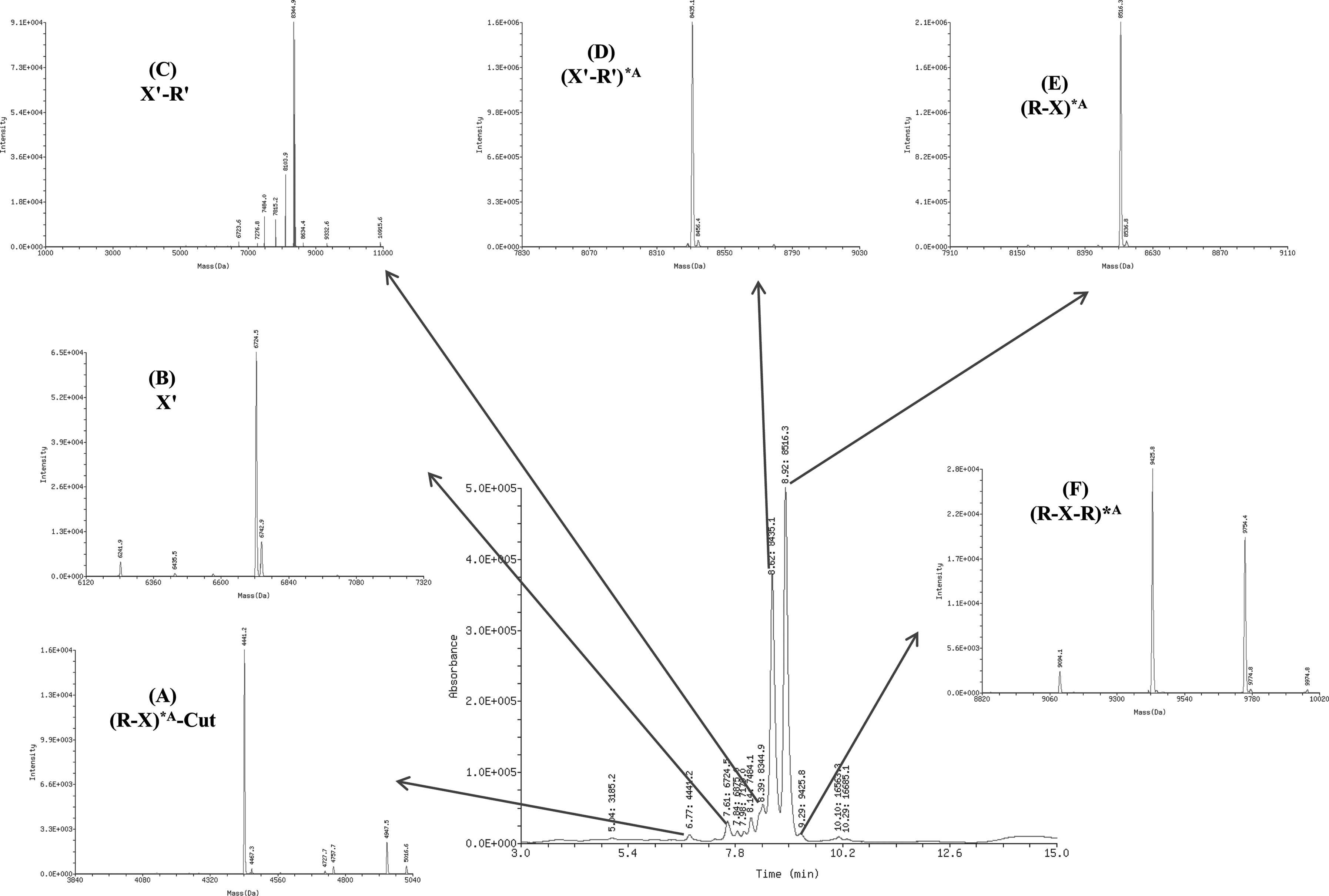
ESI-LCMS diagram of the 2′-F-dATP modified PEAR products digested by PspGI, the components
Asterisk residue (*A) represents 2′-F-dATP.
Letters with strikethrough represent the nucleotides digested by exonuclease activity of KOD DNA polymerase.
The total area percent, the sum of the sub area percents of the components, exceeded 100% because of the rounding of the summand numbers.
A, 2′-F-dATP modifications; MW, molecular weight; R, PspGI recognition site; R′, complimentary PspGI recognition site; RT, retention time; X, the target oligonucleotide sequence; X′, the complementary target sequence; underlined sequences, PspGI recognition sites.
KOD DNA polymerase has a strong 3′→5′ exonuclease/proofreading activity, which could be triggered by the modified bases incorporated in the PEAR products. Subsequently, as shown in Table 2, product E was truncated by the exonuclease activity of KOD DNA polymerase to products E-1, E-2, and E-3, and the repeat-containing products (F, G, and H) were also truncated. In Fig. 3, we also observed some low molecular weight (MW) chains, such as products A-1 and A-2. We noticed that the sequence CCTCG in the target is similar to the recognition site of PspGI (CCWGG). So A-1 and A-2 might be by-products produced by PspGI digestion by its star activity.
Fortunately, however, the proportion of truncated products is very low (4.83%), and the percentage of full-length products (B, C, D, and E) totals up to 95.17%. It is shown that the sequence of the PEAR products are maintained at an extremely high accuracy (>99.9%), and after digestion the area percent of full-length modified oligonucleotides (D and E) reaches 89.24%.
For the full-length products, since their observed MWs in mass spectrometry are fully consistent with their theoretical relative MWs, it is proved that the molecular structure of the modified PEAR products is correct.
Discussion
Synthetic antisense oligonucleotides are widely used to regulate gene expression. Undoubtedly, an antisense oligonucleotide enzymatically prepared by PEAR should have a similar biological activity, since they have a same molecular structure basically. If a double-stranded oligonucleotide is desired, in chemical synthesis, the sense and antisense strands were synthesized separately and then annealed to form duplexes. In contrast, PEAR products are already double stranded; PEAR would be a very useful technique in the production of double-stranded oligonucleotides, thanks to the high purity of the PEAR products, which will greatly simplify the purification process, and save time and cost of oligonucleotide production. However, if a single-stranded oligonucleotide is desired for in vitro or in vivo application, the PEAR products could be subjected to complete digestion and denaturing HPLC separation of the desired strand.
As high-fidelity DNA polymerases, both Phusion and KOD DNA polymerase have the ability to synthesize modified nucleotides into DNA duplexes. Comparing with unmodified dNTPs, the modifications of nucleotides help to prolong the half-life of oligonucleotides introduced into cells or improve the affinity toward targets [36]. In 2′-F-modification, the hydroxyl group in the 2′-position of furan ring is replaced by fluoro, while in S-modification, one of the two nonbridging oxygen atoms is replaced by a sulphur atom in the alpha phosphate. For thermostable DNA polymerases derived from extreme thermophilic microorganisms, unmodified dNTPs might be the most favorable substrates. When modified dNTPs are introduced in an enzymatic reaction, the substrate's affinity and the amplification efficiency will decline significantly, and sometimes even terminate. This explains the phenomenon in which some kinds of DNA polymerases (such as Taq and Pfu) are not able to incorporate modified dNTPs [37]. This can also explain the phenomenon that 2′-F-dATP and 2′-F-dGTP could not be incorporated by Phusion DNA polymerase at the same time.
In the present study, it is shown that in PEAR, KOD DNA polymerase has a broad substrate flexibility and incorporates diversified modified nucleotides. Our result is consistent with previous observations [33,38,39], which revealed that in polymerase chain reaction (PCR) KOD DNA polymerase also has higher amplification efficiency than other DNA polymerases when using modified nucleotides. It has been reported that the substrate affinity and amplification efficiency can be significantly improved by changing the specific amino acid near the substrate binding position [39]. In view of this, it is possible to change the structure of the substrate binding site of the KOD DNA polymerase to generate engineered enzymes by using point mutation, DNA shuffling, or protein evolution methods. It would be a promising strategy for the screening of DNA polymerase compatible with a wider spectrum of modified dNTPs in PEAR.
Restriction endonucleases (REases) such as EcoRI and BamHI are enzymes that cut DNA at or near specific recognition nucleotide sequences known as restriction sites [40]. Currently, >20,000 REases are listed on REBASE (http://rebase.neb.com) [41]. Among these REases, most of whose optimum temperature are 37°C, a small number of them have the ability to be resistant above 55°C, which makes them suitable for use in various applications performed at high temperatures, such as PCR. At the same time, the substrates of REases can be not only normal DNA, but also DNA with modifications. And this article might be the first report of REases cleaving DNA with different kinds of modifications in one molecule.
As we have discussed in previous publications, PEAR is simple, efficient, and stable, providing a robust alternative method for the preparation of modified oligonucleotides. PEAR products can be used directly as seeds for the next round of PEAR without any treatment, and since there is no need to add primers in multiple rounds of PEAR; this process can be repeated until sufficient amount of products is produced. Compared with traditional chemical synthesis, PEAR-based enzymatic reactions for oligonucleotide production have advantages such as being low cost, pollution free, and high purity and avoiding failure sequences. Thus, PEAR could be a promising method of choice for the large-scale production of modified oligonucleotide drugs.
Based on other studies and our experiments, we draw the conclusion that PEAR is capable of amplifying not only natural oligonucleotides[24], but also thio-[25] fluoro-modified and thio/fluoro-double modified oligonucleotides, which would meets the demand of diversified oligonucleotide production. Except for phosphorothioate and 2′-fluoro, many other kinds of modifications, including 2′-O-methoxyphenyl (MOE), 2′-methyl (OME), and locked nucleic acid (LNA), have been widely used in biomedical studies. We are further optimizing the PEAR reaction and validate the PEAR technology for the production of a variety of modified oligonucleotides.
Footnotes
Acknowledgments
This research was supported by the National Science Foundation of China through grants 81072567 and 30600463, and partially by Shandong Provincial Science Foundation grant ZR2010HM056. The funders had no role in study design, data collection, and analysis; decision to publish; or preparation of the manuscript.
Author disclosure statement
No competing financial interests exist.