
Abstract
Altering endogenous genes in cells is an integral tool of modern cell biology. The ease-of-use of the CRISPR/Cas9 system to introduce genomic DNA breaks at specific sites in vivo has led to its rapid and wide adoption. In the absence of a DNA template, the lesion is repaired by nonhomologous end joining resolving as internal deletions. However, in the presence of a homologous DNA template, homology-directed repair occurs with variable efficiencies. Recent work has demonstrated that highly efficient gene targeting can be induced by combining CRISPR/Cas9 targeting of genomic loci with recombinant adeno-associated virus (rAAV) to provide a single-stranded homologous DNA template. Here we review the current state of CRISPR/Cas-based gene editing and provide a practical guide to applying the CRISPR/Cas and rAAV system for highly efficient, time- and cost-effective gene targeting.
Short History of Gene Editing and CRISPR Biology
I
In parallel to the field of gene editing, the molecular biology of CRISPR (clustered regularly interspaced short palindromic repeats) has opened up entirely new genome-editing possibilities. Soon after their initial description in the late 1980s by Ishino et al. [19], it was realized that these sequences are made of viral nucleotide sequences and have CRISPR-associated (cas) genes in proximity [20–27]. This work resulted in the hypothesis of CRISPR/Cas being a bacterial immune system [26,28]. Within the following years, the molecular mechanisms were determined and a type II CRISPR/Cas system was identified that cuts DNA in a programmable and sequence-specific manner [29]. The identification of trans-activating crispr RNAs (tracrRNAs) and their involvement in crRNA maturation by RNase III closed the gap between missing endoribonucleases and the maturation of crRNAs [30]. Until then, all CRISPR/Cas systems had been thought to be a multiprotein/RNA system that was difficult to assemble in vitro. However, in 2011, the work of Sapranauskas et al. [31] and that of Jinek et al. [32] demonstrated that a single cas gene (Cas9) is sufficient for a fully functional type II defense system and that Cas9 is an RNA-guided DNA endonuclease [33], respectively.
The use of CRISPR/Cas for sequence-specific gene editing has since been demonstrated in a variety of cell types and tissues in eukaryotes [34–37]. The simplicity of this technology is demonstrated by an explosion of scientific literature during the last 2 years, which includes characterization of off-target mutagenic effects [38–40], improved single-guide RNA (sgRNA) design [41–43], the activation or repression of endogenous genes [44,45], the development of Cas9 nickases or dCas9-FokI fusion proteins [46–49], genome-wide knockout libraries [50–53], CRISPR/Cas9 knockin mice [54,55], and mutation of genes in a living animal [56–58]. Taken together, these recent rapid advances in our understanding of the CRISPR/Cas9 system are indicative of its bright potential for gene therapy and utilization to uncover new genetic pathways governing cell biology.
Selecting the “Right” Cas9 for Each Application
CRISPR/Cas systems can broadly be classified into three specific types (I–III) based on the combination of Cas proteins present in the genome of the respective organism [59]. The commonly used type II system is characterized by its single CRISPR ribonucleoprotein complex (Cas9), while types I and III are multiprotein complexes comprising many different Cas subunits. Interestingly, types I and II target DNA while type III has evolved to target RNA [59,60]. The fact that type II systems contain a single Cas gene has made them ideal for a variety of applications, most notably genome engineering for the purpose of “disease in a dish” models, whole animal model generation, and drug development [61].
The type II ribonucleoprotein (Cas9) is a multidomain RNA-guided DNA endonuclease with two catalytic centers, HNH and RuvC [32,62]. Two amino acid residues are of particular importance, D10 (RuvC domain) and H840 (HNH domain). Mutation of either residue to alanine converts Cas9 into a nickase enzyme with only one active catalytic center [47,48] (Fig. 1). The mutation of both residues will generate a Cas9 enzyme without DNA nuclease activity, while maintaining its ability to selectively bind DNA in an RNA-guided manner.
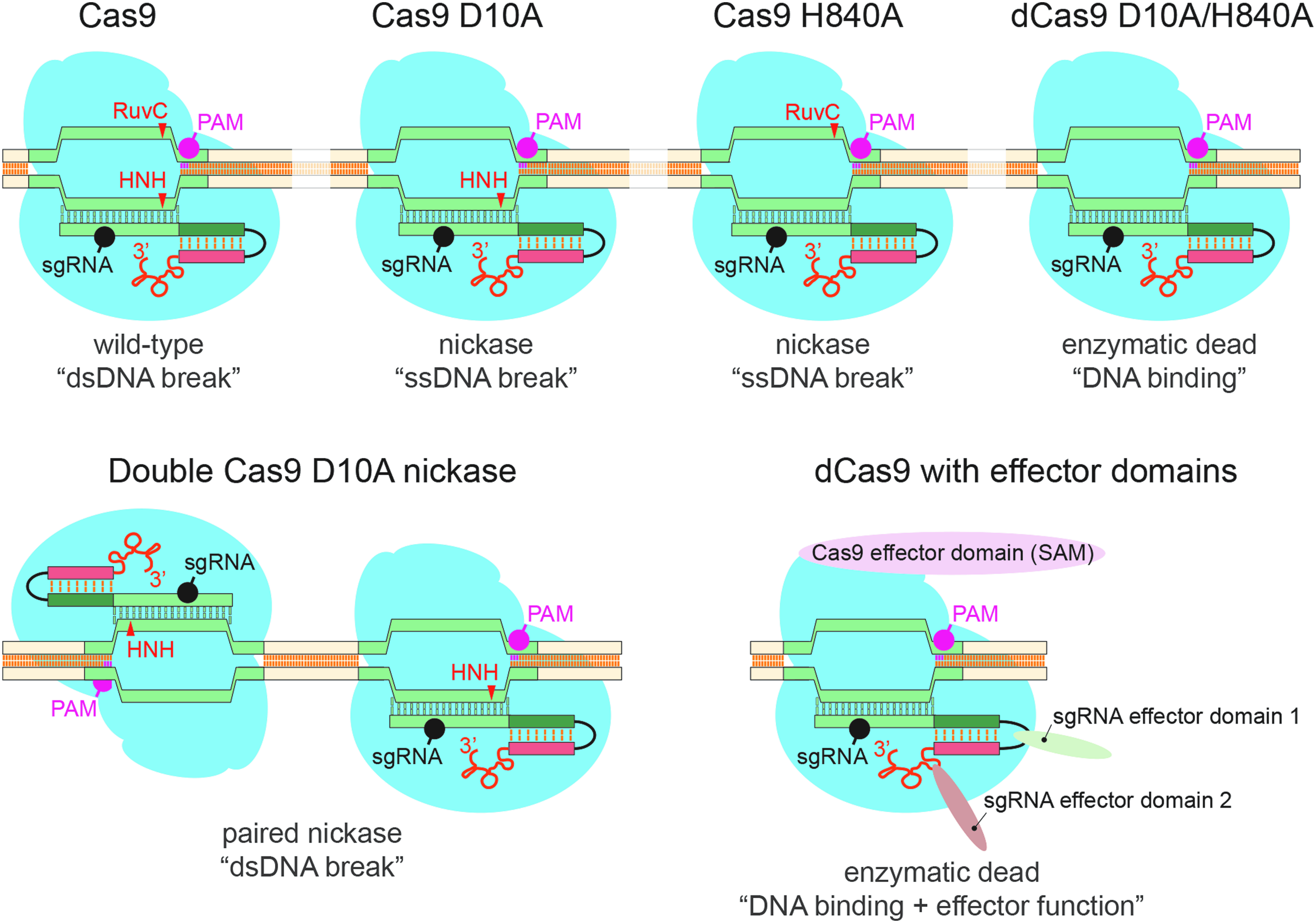
Available Cas9 species and their applications. Highlighted are the protospacer adjacent motif (PAM), the two Cas9 nuclease domains, RuvC and HNH, and the DNA position (arrowhead) on which they act as well as the position of the single-guide RNA (sgRNA). Mediated by the two nuclease domains, the wild-type enzyme generates a DNA double-strand break. Mutating a single nuclease domain, to either D10A or H840A, will result in a nickase enzyme that is only able to nick DNA at the sgRNA-targeted site. Nickase enzymes can be used in a paired manner to improve specificity and reduce off-target effects. The mutation of both nuclease domains results in a catalytically “dead” (dCas9) enzyme that retains RNA-guided DNA binding capacity. Combining dCas9 with either Cas9-linked or sgRNA-linked effector domains can be used for transcriptional activation, repression, or genomic visualization.
Wild-type Cas9 can be used as a single enzyme for indel generation, DNA insertions and replacements, as well as for gene editing, or when using multiple Cas9-gRNA complexes, for the deletion of large DNA sequences or the induction of DNA rearrangements (Fig. 1). Using the nickase as a single enzyme results in a single-stranded DNA nick, with lower efficiencies for indel generation, DNA insertions, or replacements than the wild-type Cas9 complex, but with a lower likelihood of inducing off-site mutations. This lower efficiency can be overcome by targeting two nickase complexes to the same genomic region with two different guide RNA sequences on opposing DNA strands. The resulting double-strand DNA break restores the efficiencies for indel generation, DNA insertions and replacements, and gene editing (Fig. 1) [46–49], while maintaining a low off-target effect.
The Cas9 double-mutant (dCas9; D10A, H840A) has no nuclease activity but retains its ability to bind DNA in an RNA-guided manner, making it useful for other applications. Fusing dCas9 N- or C-terminal to effector domains allows for gene activation (VP64 domain) [45,63,64], repression (KRAB domain) [45,63], visualization (fluorescent proteins) [65–67], or chromatin modification (histone modifier or DNA methylation regulators) [68] (Fig. 1). Interestingly, an sgRNA sequence is often not sufficient to drastically change the expression profile of a gene, instead a pool of up to five RNA guide sequences is often required to target multiple complexes to the same region. To circumvent this obstacle, structure-guided engineering of the CRISPR/Cas9 complex produced to an internal VP64 domain with additional RNA aptamer sequences into the gRNA that facilitate the recruitment of aptamer-binding domain-fused activator domains to the CRISPR/Cas9 complex [69]. This results in a dCas9-VP64:sgRNA:aptamer-effector complex that activates gene promoter transcription very efficiently with an sgRNA (Fig. 1).
How to Choose a Guide RNA?
The most difficult task in the process of applying CRISPR/Cas9 technology is identifying the “best” guide RNA sequence. To increase the likelihood of selecting a highly efficient guide RNA, a few selected parameters need to be considered. The first is choosing the position of the guide sequence with respect to the region of interest (promoter, enhancer, coding region, untranslated region, miRNA, etc.), which depends on the intended application (Fig. 2A). For transcriptional activation, the guide sequence should be 450 to 50 nucleotides upstream of the transcriptional start site (TSS), whereas for transcriptional repression it should be between 50 and 450 bases downstream of the TSS [45]. To generate indel mutations for the purpose of gene disruption, the guide sequence should ideally be located within the first three exons of the gene of interest [41]. However, recent work demonstrated that guide RNAs targeting functional protein domains have a higher probability to induce gene-function disruption than other guide RNAs [70]. For gene editing in the form of introducing coding missense mutations or gene tagging, the guide sequence should be located within an adjacent intron or noncoding region to avoid inadvertent disruption of the other allele [37].
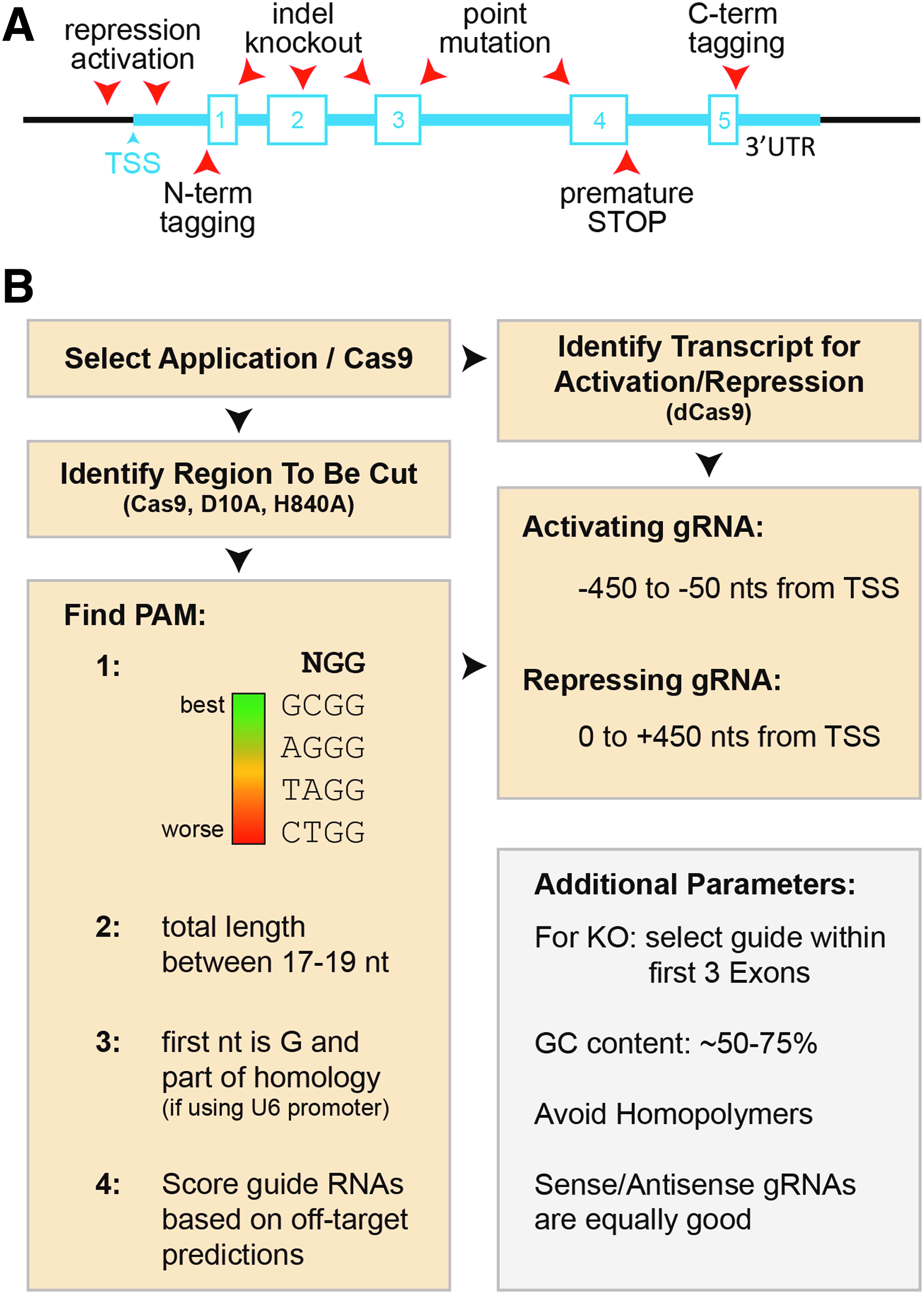
A guide to selecting a guide RNA.
Once the approximate genomic target region has been selected, identification of a specific guide sequence takes advantage of the insights gained from high-throughput studies that have identified the main parameters affecting the quality of guide sequences (Fig. 2B) [41,42,45,71]. First, and most importantly, is the identification of a protospacer adjacent motif (PAM) sequence. For simplicity, we focus on PAM sequences of the NGG format for Cas9 from Streptococcus pyogenes, the most widely used Cas9. Characterization of 1,841 coding DNA sequence-targeting guide sequences revealed that PAMs of the GCGG format have the highest probability of success, whereas gRNAs utilizing the PAM sequence CTGG are the least likely to function [41]. In terms of efficiency and off-target effects, the use of truncated guide sequences has been shown to keep on-target efficiencies high, but dramatically reduce off-target rates. Therefore, the length of the guide RNA recognizing the DNA of interest should be kept to 17–19 nts [42]. Also, the GC content should ideally be between 50% and 75% and the presence of homopolymers (GGGG, CCCC, AAAA, TTTT) should be avoided [45]. Interestingly, guide RNAs located on the sense or antisense strand work with similar efficiencies [45].
Finally, once a guide RNA sequence has been chosen, the sequence can be examined for potential off-target annealing sites by analyzing the sequence with published algorithms [41,71]. Knowing that the CRISPR/Cas system is a bacterial immune system and that the average size of a bacterial genome is 3–5 Mbp compared to the size of the human genome at 3 billion base pairs [72–74], the potential off-target cleavage effects of guide RNA sequences are increased by a factor of 1,000. Therefore, it is important to highlight that no matter how “good” the selected guide sequence is, every guide sequence will have off-target effects when used in mammalian cells.
Recombinant Adeno-Associated Virus as an Optimal Knockin Template
Two prerequisites are required for efficient CRISPR/Cas9 gene targeting in cells. First, a DNA double-strand break within the vicinity of the desired region. Second, a DNA template needs to be provided that can induce HDR instead of NHEJ. Different homology-directed templates have been described, among them single-stranded oligonucleotides (ssDNA) of up to 200 nts length [75,76], double-stranded circular or linearized plasmid DNA (dsDNA) with homology arms of varying length, including bacterial artificial chromosomes [77], and recombinant adeno-associated virus (rAAV) single-stranded DNA of up to 4.5 kb length [37] (Fig. 3). While short ssDNA oligonucleotides have the advantage of rapid design and generation, their disadvantages are the requirement of nucleofection for cellular delivery, poor recombination frequencies, and the inability to include selectable markers, resulting in time-consuming clonal selection and screening downstream. In contrast, long dsDNA templates with large homology arms allow the delivery of resistance genes for clonal selection. However, dsDNA has the major disadvantage of being a very poor substrate for HDR, as well as its ability to randomly integrate into the genome by NHEJ (Fig. 3). In contrast to oligonucleotides, single-stranded rAAV DNA templates retain the advantages of long homology arms and encoding resistance genes for increased recombination efficiencies with low NHEJ rates by being the preferred template for HDR, leading to high targeting frequencies [78]. However, rAAV vectors can integrate at sites of low homology in an NHEJ-dependent manner, but the process is far from being random [79]. Factors determining the rate of “random” integration include cell type and genomic sites. Interestingly, 3%–8% of all “random” integration events happen in ribosomal DNA repeats [80,81]. In addition, CpG islands and the TSS (±1,000 bp) were identified as preferred “random” integration sites [82]. Although no explanation has been given for this “nonrandom” event, it is likely that the high-CG content (>70%) of the rAAV is driving these events. To identify NHEJ-dependent rAAV integration, Southern blot, polymerase chain reaction (PCR), fluorescence in situ hybridization analysis [83,84], or rAAV shuttle vectors were used to identify the sequence of the vector:chromosome junctions [85,86].
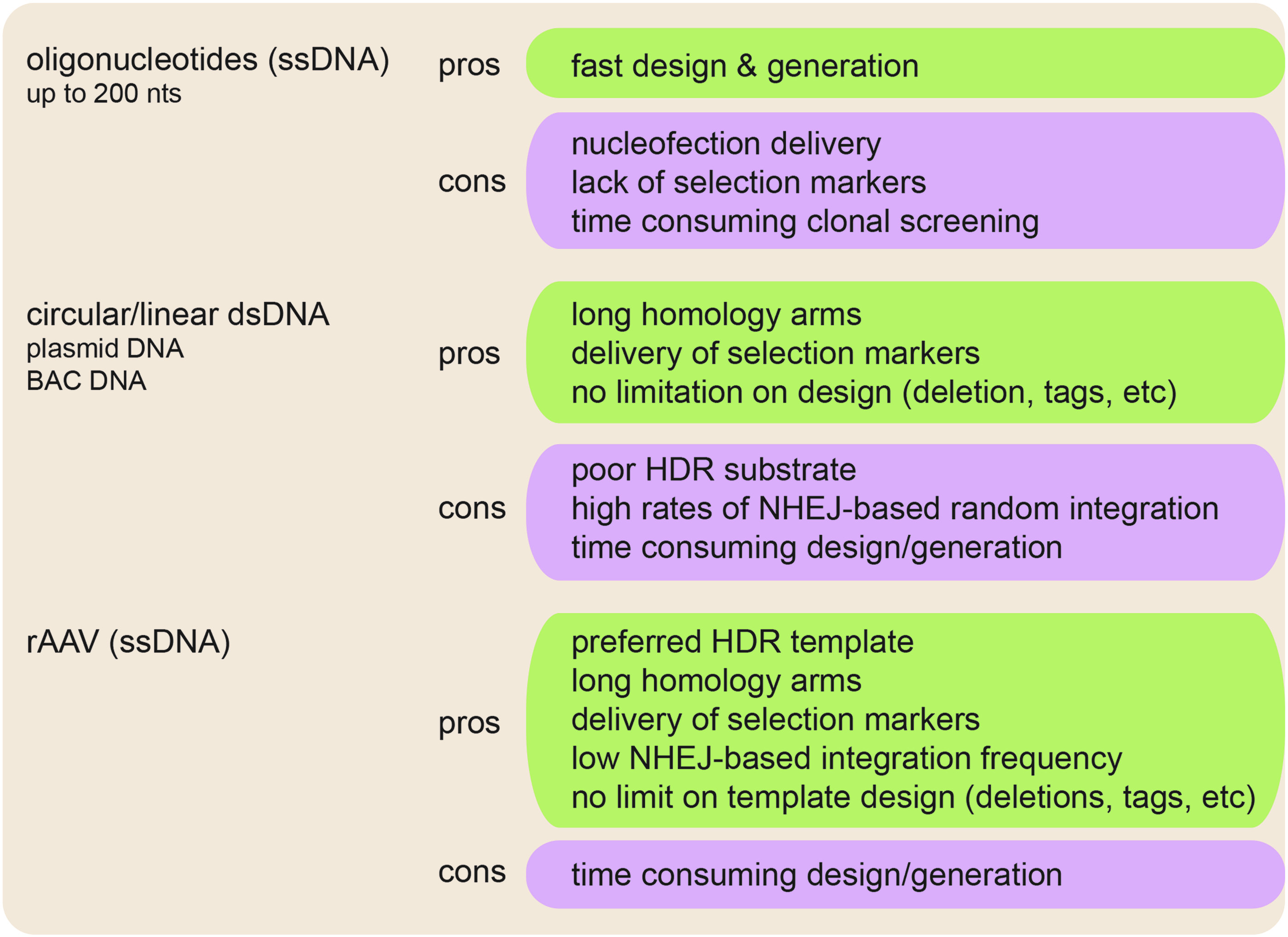
Advantages and disadvantages of homology-directed repair (HDR) templates. Oligonucleotides of up to 200 bases length, circular or linear plasmid DNA, and single-stranded DNA in the form of recombinant adeno-associated virus (rAAV) are the most frequently used HDR templates. Listed are positive (green box) and negative (purple box) features of each template type.
rAAV as a DNA delivery vehicle
AAV is a single-stranded DNA virus of the Parvoviridae family. AAVs, independent of the serotype, are small icosahedral viruses with a single 4.7-kb DNA genome that contains hairpin-shaped inverted terminal repeats at the 5′ and 3′ ends. AAV contains two open reading frames, rep and cap, for nonstructural and structural genes, respectively. By deleting the rep and cap genes and replacing them with transgenic mammalian sequences and providing the cap gene in the form of a cotransfected plasmid, rAAV can be generated in cell culture for gene-editing purposes. Nine AAV serotypes have been reported (AAV1–9), with AAV2 being the most commonly found in humans (80%). In addition, a chimeric serotype (AAV-DJ) has been engineered in vitro with a capsid protein comprising a 60-amino acid hybrid derived from types 2/8/9 that outperform most natural serotypes in cell culture [87]. According to NIH guidelines, serotypes 1–4, and all recombinant rAAV constructs that do not contain potentially tumorigenic gene products or toxin molecules and that are produced in the absence of a helper virus, can be handled at biosafety level 1, making rAAV a widely useful reagent for gene editing with low to no biosafety concerns.
Selection Cassettes
Depending on the desired gene-targeting event, there are multiple ways to design an rAAV template for HDR. Figure 4 illustrates the most commonly used targeting strategies, namely, the introduction of coding missense mutations, premature stop codons, and N- and C-terminal tagging of genes. Among others, we discuss two different approaches to introduce coding missense mutations that differ in their respective template design. The first uses a splice acceptor (SA) site linked to an internal ribosomal entry site (IRES) and a selection gene [88], either an antibiotic resistance gene or fluorescence reporter for subsequent clonal selection or single cell sorting, respectively, which results in an extremely high gene-targeting frequency of 80%–90% of clones [37]. This strategy is particularly useful if high efficiencies are required.
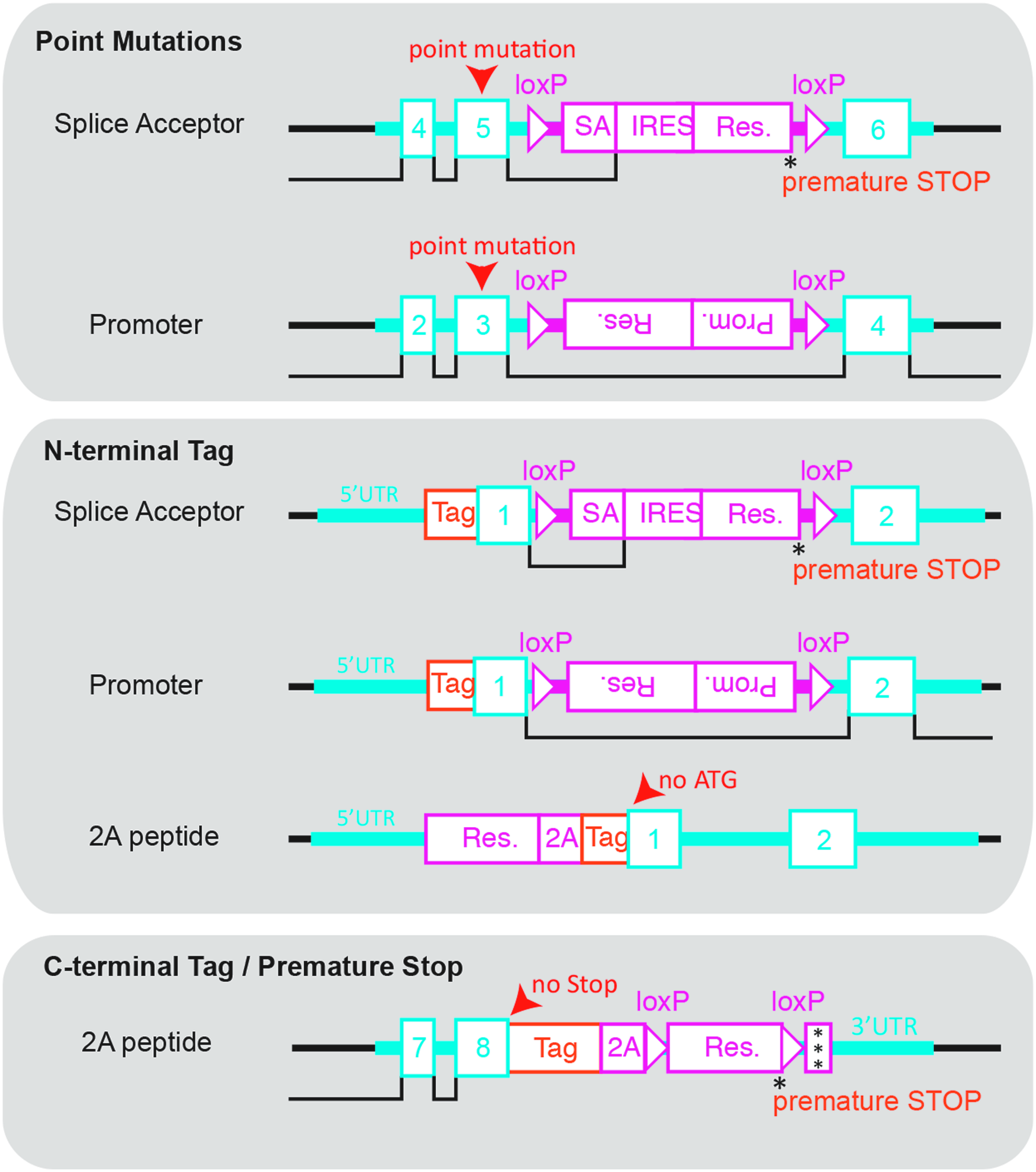
HDR template design strategies. The complete rAAV HDR template consists of left and right homology arm (light blue) containing exons (numbered boxes) and introns, as well as a selection cassette (red) that is separated from the homology arms by loxP sites for subsequent Cre recombinase excision of the selection cassette. Point mutations (red arrowhead), tags (orange), and premature stop codons (asterisk) can directly be cloned into the homology arm to obtain the final rAAV HDR template.
However, due to premature termination of transcription at the selection marker's stop codon, it is important to highlight that integration of the selection cassette temporarily inactivates the allele. The selection cassette is flanked by loxP sites, allowing for Cre recombinase-mediated excision of the selection cassette and reactivation of the targeted allele. If essential genes are targeted with this strategy, only heterozygous clones can be obtained and a second round of gene targeting will be required to obtain clones homozygous for the desired mutation [37]. If the essential gene is haploinsufficient, an alternative strategy needs to be applied. In this case, an inverse cassette comprising a promoter and selection gene can be introduced into an intron adjacent to the site of interest. This strategy avoids the temporal inactivation of the allele but has the disadvantage of delivering a promoter-driven selection, which in the case of off-site template integration will result in a higher incidence of viable false-positive clones and a more time-consuming clone selection process. Similar to the SA-IRES cassette, the promoter-driven selection cassette can be excised by Cre recombinase.
One very useful gene-targeting application is the introduction of N- or C-terminal high avidity epitope or fluorescent tags. Different strategies are required for N-terminal versus C-terminal tagging. Similar to the introduction of point mutations, the introduction of N-terminal tags uses either an SA-IRES or inverted promoter for resistance gene expression (Fig. 4). The major difference compared to the introduction of point mutations is that the desired tag is fused to the first exon during the process of homology arm generation. Despite that difference, N-terminal tags and point mutations use a similar template design. In addition, a new selection cassette design has been proposed, but waits for laboratory testing. In this new design, the left homology arm, which encodes the endogenous promoter, drives a resistance gene that is separated from exon 1 of the gene of interest by a 2A sequence and the desired tag sequence (Fig. 4). Combined with a 5′ or 3′ death cassette such as diphtheria toxin A, this approach, theoretically, reduces the rate of false-positive clones significantly by maintaining the rate of true-positive clones. Probably the biggest advantage of this design is the fact that the tag sequence is not considered a break in homology, making it feasible to integrate even large tags such as fluorescent proteins. In contrast, introduction of C-terminal tags yields high targeting rates without having to resort to an SA-IRES or a promoter to drive expression of the resistance gene. Instead, the last exon, which is contained in the left homology arm, is fused in frame to the desired tag—thereby removing the endogenous stop codon. The tag is followed by an in-frame 2A peptide sequence [89], a loxP site, and the selection gene for antibiotic selection. Another loxP site followed by a triple stop codon sequence completes the selection cassette (Fig. 4). It is noteworthy that this strategy does not inactivate the allele, making it possible to target essential genes without affecting cell viability. Like the previous template designs, the selection cassette can be excised by Cre recombinase treatment.
Designing Homology Arms
Careful design of the left and right homology arms of the template is essential for successful gene targeting. In general, longer template DNA will result in higher targeting efficiency [90]. Also, if the template DNA contains breaks in homology, for example, missense mutations, tags, or the selection cassette, the homology break should be designed to be centered within the overall template [90]. Because the endogenous AAV genome is 4.7 kb and is packed with maximal efficiencies by the capsid proteins, the length of the rAAV template should be close to 4.5 kb and never exceed 5.2 kb. Templates exceeding 5.2 kb will be truncated at the 5′ end during the packaging process, resulting in a heterogeneous rAAV particle population with reduced transgene delivery efficiencies [91]. In sum, the overall rAAV template size should be designed around 4–4.5 kb with breaks in homology as close to the center of the template as possible.
In addition to these general rules, here are some empirically derived guidelines. First, the 5′ and 3′ ends of the left and right homology arm should, if possible, start and end within noncoding regions, respectively. Although the process of HDR is highly efficient and mostly scarless, this design avoids the potential introduction of coding region errors during the recombination process. To make the PCR-based identification of faithfully recombined clones during the clone selection process as sensitive as possible, one homology arm should be kept less than 1 kb, and the other arm to be made as long as necessary to “fill up” the rAAV. To avoid single nucleotide breaks in homology, all homology arms should be amplified using proofreading polymerases from genomic DNA isolated from the tissue or cell line that will be used for the genome editing.
Combining CRISPR/Cas9 with rAAV Templates for Highly Efficient Gene Editing
Combined use of CRISPR/Cas with rAAV donors effects high frequency and scarless gene targeting [37]. Specifically, rAAV donors yield high on-target integration rates while providing little off-target effects, and offering precise gene editing on actively transcribed open reading frames [37]. The successful combination of both technologies depends on a few parameters. In addition to the design of guide RNA and HDR donor template, efficiently introducing the individual components into cells is very important. While the delivery of HDR templates is “limited” to single-stranded rAAV, the components of the CRISPR/Cas system can be delivered in multiple ways. Most cell types can be transfected with chimeric plasmid constructs. If this yields insufficient recombination frequencies, transfecting Cas9 mRNA [92] or whole protein can increase targeting frequency [93]. Most recently, the components of the CRISPR/Cas system have been delivered by rAAV [54,58,94]. However, how this Cas9/gRNA delivery method affects the correct recombination rate during gene editing remains to be seen.
Concluding Remarks
Performing gene editing in human cells to generate disease models, to recapitulate the effect of known mutations in vitro, or to study protein function with mutant protein expression levels at physiologic levels remains a major challenge. Initial work with rAAV demonstrated its potential as an HDR template, but low targeting efficiencies prevented the technology from being widely used in cell biology laboratories. The underlying reason for the low targeting efficiency is the requirement of a DNA double-strand break in proximity of the targeted region that induces HDR. The CRISPR/Cas9 system closes this gap by enabling researchers to specifically target DNA and induce DNA breaks wherever a PAM sequence is present. Indeed, the combination of both technologies has been shown to result in high gene-targeting rates for heterozygous and homozygous targeting strategies [37]. However, several open questions remain: (1) How to identify and validate off-target effects? (2) How to avoid the CRISPR/Cas9-mediated cut on the second strand for heterozygous targeting strategies? (3) How to increase the overall targeting efficiency with HDR templates that do not contain selectable markers?
First, how to identify off-target effects, two methods are commonly used: (1) the use of “off-target” prediction tools with subsequent conventional sequencing and TIDE analysis [95], Surveyor or T7 endonuclease I (T7EI) assays [34,38], and (2) deep sequencing on predetermined target sequences [39,56,96,97]. While Surveyor and T7EI assays can be routinely performed on a selected set of genomic regions, it is not feasible to perform these assays genome wide. In contrast, whole genome (deep) sequencing can identify all potential off-target effects, but it is not cost-efficient enough to be performed on a regular basis at this point in time. However, methods such as GUIDE-seq, Digenome-seq, or BLESS are potential tools for the unbiased detection of off-target cleavage by CRISPR/Cas9 [43,98,99]. In addition, combining CRISPR/Cas9 and rAAV HDR templates makes the generation of gene-recombined resources easy and fast, demanding methods other than deep sequencing to assess all potential off-target effects. In addition to CRISPR/Cas9-mediated off-target effects, the HDR template can induce off-target random integration in an NHEJ-dependent manner. As expected, these off-target effects are more difficult to dissect since the integration is more or less random. However, selected assays can be used to identify this effect. The most widely used approach is Southern blot analysis [100]. Another approach is represented by PCR-based detection of the HDR template in predicted guide RNA off-target sites [101], although it is less comprehensive.
Second, targeting a single copy of a gene with CRISPR/Cas9 is challenging. Indeed, when gene-targeting strategies are used to mutate a single copy of an allele, the second copy, even if not recombined by rAAV, is very likely to be targeted by CRISPR/Cas9, resulting in indel formation with potential phenotypic consequences. While the distinction between heterozygous and homozygous allele targeting can be made by performing allele-specific PCRs, limiting the presence of the CRISPR/Cas9 components may help to solve this problem. In fact, the use of a doxycycline-inducible CRISPR/Cas9 system regulates the frequency and size of target gene modifications [55]. Alternative CRISPR/Cas9 component delivery methods, in the form of protein [102,103], mRNA [103,104], or as split Cas9 [105,106], may also improve the temporal control over Cas9 activity.
The use of selectable markers in rAAV HDR templates has been shown to result in extremely high gene-editing rates of selectable clones. However, relative to the total number of cells that need to be transfected and transduced with the CRISPR/Cas9 components and the rAAV template, the rate is shockingly low. Given the fact that there are four populations of cells during those experiments (CRISPR/Cas9 positive, rAAV template template positive, and double positive and double negative) and only the double-positive population contains the cells that can undergo faithful recombination, the following question remains: how to increase the double-positive population? One interesting possibility may be the use of rAAV, not only to deliver the HDR template but also to deliver the Cas9 components. Cas9 and gRNA delivery by rAAV have recently been shown to work very effectively in whole animals by direct injection [54,58]. However, whether this approach, together with rAAV template delivery, results in higher “overall” gene-targeting rates without the use of selectable markers remains to be tested. All told, CRISPR/Cas9 systems have turned what was previously a specialist-only field of genome editing into a commonly used molecular cell biology approach on par with PCR and RNAi.
Footnotes
Acknowledgments
We are thankful to A. Springer, A. Kacsinta, and S. Heinz for critical comments. The preparation of this article was supported, in part, by grants from the NIH (CA185589).
Author Disclosure Statement
No competing financial interests exist.