
Abstract
We report the effect of introducing a single incorporation of 2-thio-deoxythymidine (2S-dT) or C5-Triazolylphenyl-deoxythymidine (5-TrPh-dT) at four positions within the gap region of RNase H gapmer antisense oligonucleotides (ASOs) for reducing wild-type and mutant huntingtin mRNA in human patient fibroblasts. We show that these modifications can modulate processing of the ASO/RNA heteroduplexes by recombinant human RNase H1 in a position-dependent manner. We also created a structural model of the catalytic domain of human RNase H bound to ASO/RNA heteroduplexes to rationalize the activity and selectivity observations in cells and in the biochemical assays. Our results highlight the ability of chemical modifications in the gap region to produce profound changes in ASO behavior.
Introduction
H
Several oligonucleotide-based approaches have been investigated as potential allele-selective and allele-nonselective HD therapeutics [5]. We recently described second-generation antisense oligonucleotides (ASOs) targeting SNP rs7685686_A in intron 42 of the huntingtin (HTT) gene for allele-selective suppression of mu HTT [6]. SNP rs7685686_A is tightly linked to the CAG expansion, and ASOs targeting this SNP can provide an allele-selective therapeutic option for roughly half of the Caucasian HD population. Second-generation gapmer ASOs are typically fully phosphorothioate (PS) [7]-modified chimeric oligonucleotides with a central DNA gap region flanked on either end with affinity-enhancing nucleotides [8]. Gapmer ASOs bind to their cognate mRNA in cells and promote RNA degradation through RNase H1-mediated hydrolysis [9]. RNase H1 is a ubiquitously expressed enzyme, which selectively cleaves the RNA strand of a RNA/DNA heteroduplex [10,11].
The starting point for our studies was the previously identified ASO A1, which is fully complementary to the mu HTT mRNA, but has a TG mismatch to the wt HTT mRNA at position 8 corresponding to the SNP site (Fig. 1) [12]. This ASO has a 9-base DNA gap flanked on either end with 2′,4′-constrained-2′-O-ethyl bridged nucleic acid (cEt) [13] and 2′-O-methoxyethyl RNA (MOE) nucleotides [14]. We showed that replacing deoxythymidine (dT) at position 5 of ASO A1 with 2-thio-deoxythymidine (2S-dT) produced a profound enhancement in allele selectivity, while analogous substitution at position 6 did not [12]. Interestingly, ASOs modified with 2S-dT differed from A1 only in the replacement of one oxygen atom with sulfur at the C2 position on the pyrimidine nucleobase. This suggested that very small structural changes on the nucleobases in the ASO can have a profound impact on the RNase H activity.
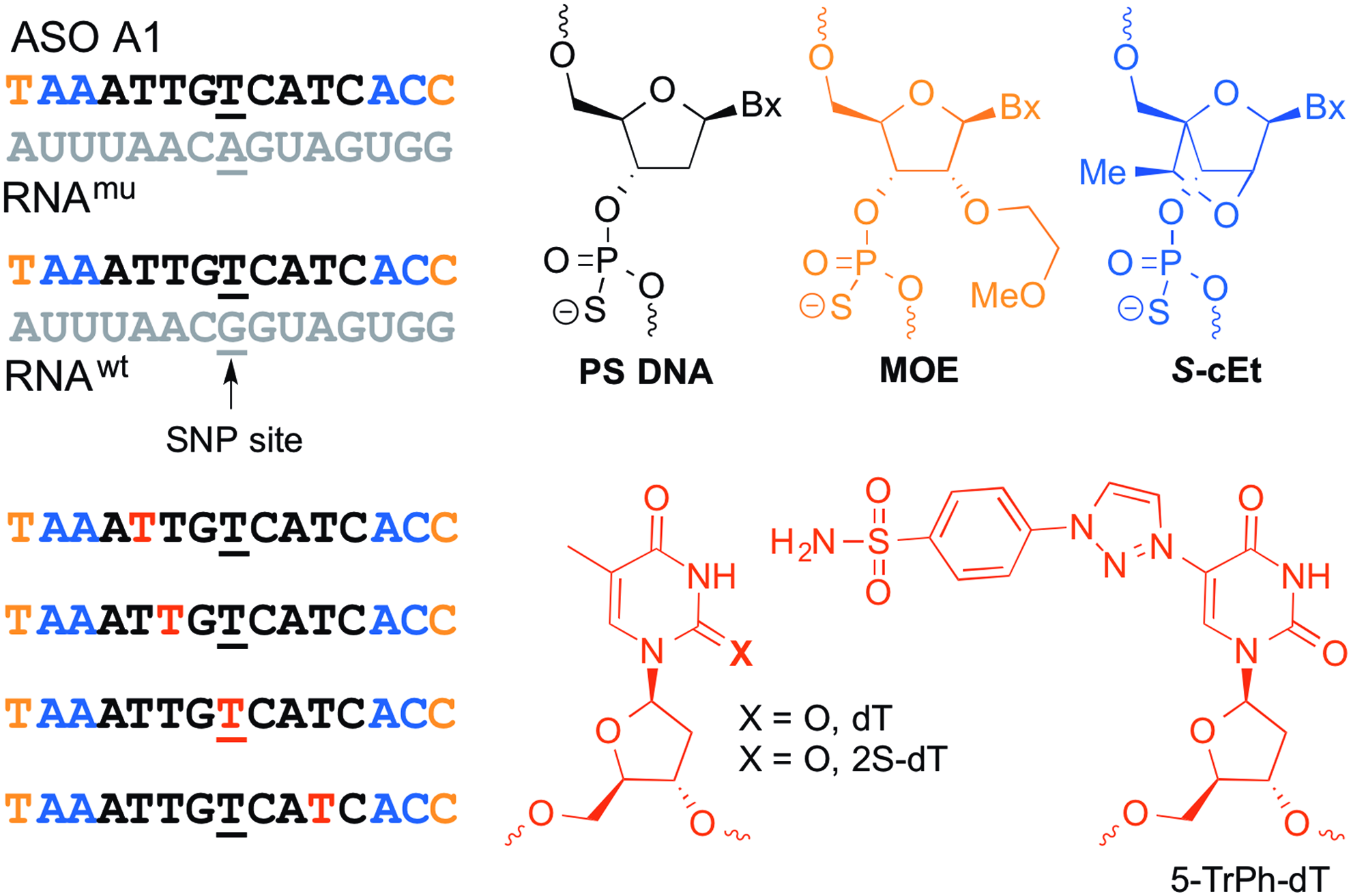
Sequence of parent ASO A1 and of the complementary RNA strands representing the wild-type and mutant HTT mRNA. The black letters indicate DNA, orange MOE, blue cEt, and red letters show site of replacement of 2′-deoxythymidine (dT) nucleotides in the gap of parent ASO A1 with 2S-dT or 5-TrPh-dT. Underlined T indicates nucleotide across from the SNP site. All ASOs are fully phosphorothioate backbone modified. ASO, antisense oligonucleotide. Color images available online at www.liebertpub.com/nat
The C5-position on the pyrimidine nucleobases is another position amenable for substitution, and C5-methyl-substituted pyrimidines represent the most widely used nucleobase modification in antisense therapeutics [15]. The 5-methyl groups enhance nuclease stability and increase duplex thermal stability (Tm) by stacking between the heterocyclic nucleobases in oligonucleotide duplexes [16]. In addition, 5-methyl substitution on deoxycytidine nucleotides greatly suppresses the immunostimulatory profile of CpG oligonucleotides [17].
Replacing the 5-methyl group on the pyrimidine nucleobase with other substituents such as 5-alkynyl [18–22], 5-thiazolyl [23], or 5-heteroaryl [23] further enhanced duplex thermal stability and nuclease stability. Elaboration of these designs has recently led to the synthesis of 5-triazole-substituted pyrimidine DNA analogs, which can provide dramatic enhancements in duplex thermal stability when positioned sequentially within an oligonucleotide as a result of stacking of the aromatic moieties in the major groove [24–26]. In these designs, 2′-deoxythymidine with bulky 5-triazolylphenyl bearing a distal sulfonamide moiety substituted (5-TrPh-dT, Fig. 1), produced the highest increase in duplex thermal stability. However, the biological evaluation of oligonucleotides modified with 5-TrPh-dT monomers has never been reported. Furthermore, 5-TrPh-dT presented an opportunity to compare the effects of introducing a large substituent in the major groove versus substitution on the Watson–Crick base pairing face (2S-dT) of DNA/RNA heteroduplexes, on the activity and selectivity profile of SNP targeting ASOs.
In this article, we report the effect of a single incorporation of 5-TrPh-dT or 2S-dT at four positions in the gap region of RNase H ASOs targeting a SNP in the HTT mRNA. We show that single incorporation of these modifications is well tolerated and can provide meaningful improvements in selectivity of SNP targeting ASOs for allele selective suppression of gene expression. In addition, we present a model that provides a structural rationale for the biological results.
Materials and Methods
Oligonucleotide synthesis
Oligonucleotides on a 2 μmol scale were made on an ABI 394 DNA/RNA synthesizer using MOE 5-methyl-C Primer support (loading 215 μmol/g). Fully protected nucleoside phosphoramidites were incorporated using standard solid-phase oligonucleotide synthesis, that is, 3% dichloroacetic acid in dichloromethane for deblocking, 1 M 4,5-dicyanoimidazole 0.1 M N-methylimidazole in acetonitrile as an activator for amidite couplings, acetic acid in THF and 10% 1-methylimidazole in THF/pyridine for capping, and 0.2 M phenylacetyl disulfide in pyridine:acetonitrile 1:1 (v:v) for thiolation. DNA building blocks were dissolved in acetonitrile (0.1 M) and incorporated using two times the 4-min coupling time, while other building blocks were dissolved in acetonitrile:toluene 1:1 (v:v) and coupled for 2 times 6 min. After conclusion of the synthesis, the 5′ DMT group was removed and cyanoethyl protecting groups cleaved using triethylamine:acetonitrile 1:1 (v:v). The remaining protecting groups were removed in concentrated aqueous ammonia at room temperature for 12 h. ASOs were purified by ion-exchange-HPLC using a linear gradient of buffer A and B. Buffer A: 50 mM NaHCO3, buffer B: 1.5 M NaBr, 50 mM NaHCO3—both buffers in acetonitrile:water 3:7 (v:v). Purified ASOs were desalted using C18 reverse-phase cartridges and analyzed by UV and mass spectrometry (Table 1).
ASO, antisense oligonucleotide.
Thermal denaturation studies
ASO and RNA were mixed in the 1:1 ratio (4 μM duplex) in a buffer containing 10 mM phosphate, 100 mM NaCl, and 10 μM EDTA at pH 7.0. Duplexes were denatured at 85°C and slowly cooled to the starting temperature of the experiment (15°C). Thermal denaturation temperatures (Tm values) were measured in quartz cuvettes (path length 1.0 cm) on a Cary 100 UV/VIS spectrophotometer equipped with a Peltier temperature controller. Absorbance at 260 nm was measured as a function of temperature using a temperature ramp of 0.5°C per min. Tm values were determined using the hyperchromicity method incorporated into the instrument software. RNAs used were as follows:
Human RNase H1 cleavage pattern
RNA was 5′-end labeled with 32P using 20 U of T4 polynucleotide kinase, 120 pmol (7,000 Ci/mmol) of [γ-32P]ATP, 40 pmol RNA, 70 mM Tris-HCl, 10 mM MgCl2, and 50 mM DTT, at pH 7.6. The labeling reaction was incubated at 37°C for 30 min followed by heating at 90°C for 1 min. Labeled RNA was purified using 12% denaturing polyacrylamide gel. ASO (200 nM), unlabeled 19mer RNA (100 nM; same RNA as used for Tm experiments), and a small amount of 32P-labeled RNA were mixed in the hybridization buffer (20 mM Tris, 20 mM KCl, pH 7.5) and heated to 90°C for 2 min. To the hybridization mixture were added 0.1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 1 mM MgCl2, and 40 U of RNaseOUT and incubated at 37°C for 1 h. The human RNase H1 enzyme was incubated in the dilution buffer (20 mM Tris, 50 mM KCl, and 2 mM TCEP, pH 7.5) for 1 h at room temperature. The enzyme solution (1 μL/25 μL duplex solution) was added to duplex solution and incubated at 37°C. After 45 min, reactions were quenched in the loading buffer and snap-frozen on dry ice. Cleavage products were separated by 12% denaturing polyacrylamide gel and products were quantitated on a phosphorimager.
Allele-selective polymerase chain reaction conditions
GM04022 fibroblasts heterozygous at SNP rs7685686_A were used to measure the in vitro potency and selectivity of the modified ASOs. The cells were trypsinized and resuspended at a density of 260,000 cells per mL in growth medium before transfecting varying concentrations of ASOs with electroporation (Harvard Apparatus ECM830, 130 V, 6 ms). Treated cells were maintained at 37°C and 5% CO2 in a minimal essential medium containing 15% fetal bovine serum, nonessential amino acids, and penicillin/streptomycin. Approximately 24 h post-transfection, the cells were washed with Dulbecco's phosphate-buffered saline buffer and lysed. RNA was extracted using the Qiagen RNeasy96 kit, and levels of the human HTT mRNA alleles were determined using the qPCR assay C_2229297_10 at SNP rs362303 from Life Technologies. The mu and wtHTT mRNA levels were measured simultaneously using two different fluorophores, FAM for the mutant allele and VIC for the wild-type allele. Quantitative real-time polymerase chain reaction (RT-PCRs) were run on the ABI 7900HT instrument using the QuantiTect Probe RT-PCR Kit following the manufacturer's instructions. The HTT mRNA levels were normalized to total RNA content, as measured by RiboGreen. Data are expressed as mean±standard deviation (n=3). Sequence of the allele nonselective ASO was
Structural model
The structural model was created from the published X-ray crystal structure (pdb code 2QK9) of the catalytic domain of human RNase H1 bound to a DNA/RNA heteroduplex [27] using Discovery Studio 4.0 visualizer software. Oxygen atoms at the C2-position of pyrimidine nucleobases at relevant locations from the published models were changed to sulfur and distances to relevant amino acid side chains were mapped. The nucleobase of 5-TrPh-dT was created in ChemBio3D Ultra14.0 and used in the structural model.
Results
Effect of 2S-dT and 5-TrPh-dT on thermal discrimination of GT mismatch
The ASOs were first tested in Tm assays versus the matched RNA complement
The black letters indicate DNA, orange MOE, blue cEt, and red and green letters show site of replacement of 2′-deoxythymidine (dT) nucleotides in the gap of parent ASO A1 with 2S-dT or 5-TrPh-dT, respectively. Underlined letters indicate position across from the SNP site. All ASOs are fully phosphorothioate backbone modified. Nonselective ASO is a 5–10–5 MOE gapmer, which targets HTT mRNA at site distinct from SNP rs7685686_A. Negative control ASO is a 5–10–5 MOE gapmer, which does not target HTT. Color images available online at www.liebertpub.com/nat
NA, not applicable.
Effect of 2S-dT and 5-TrPh-dT on activity and allele selectivity in human fibroblasts
ASOs B1–B4 and C1–C4 were tested in GM04022 human fibroblasts for their ability to reduce HTT mRNA, and the reduction of the individual alleles was measured using allele-selective qRT-PCR (Fig. 2 and Table 2) [12]. In addition, we also included a nonallele-selective ASO D1, which does not bind at the targeted SNP site on the HTT mRNA and ASO E1, which does not target the HTT mRNA, as controls for the assay. As seen in our previous study, introducing 2S-dT at position 5 resulted in a profound increase in allele selectivity while maintaining potency versus the mutant allele (<20% reduction in wt HTT at 10 μM; IC50 for reducing mu HTT 0.49 μM). In contrast, introducing 2S-dT at position 6 (B2) resulted in similar potency, but reduced selectivity compared to A1. Introducing 2S-dT at positions 8 (B3) and 11 (B4) showed similar potency and selectivity profile as the parent ASO A1.
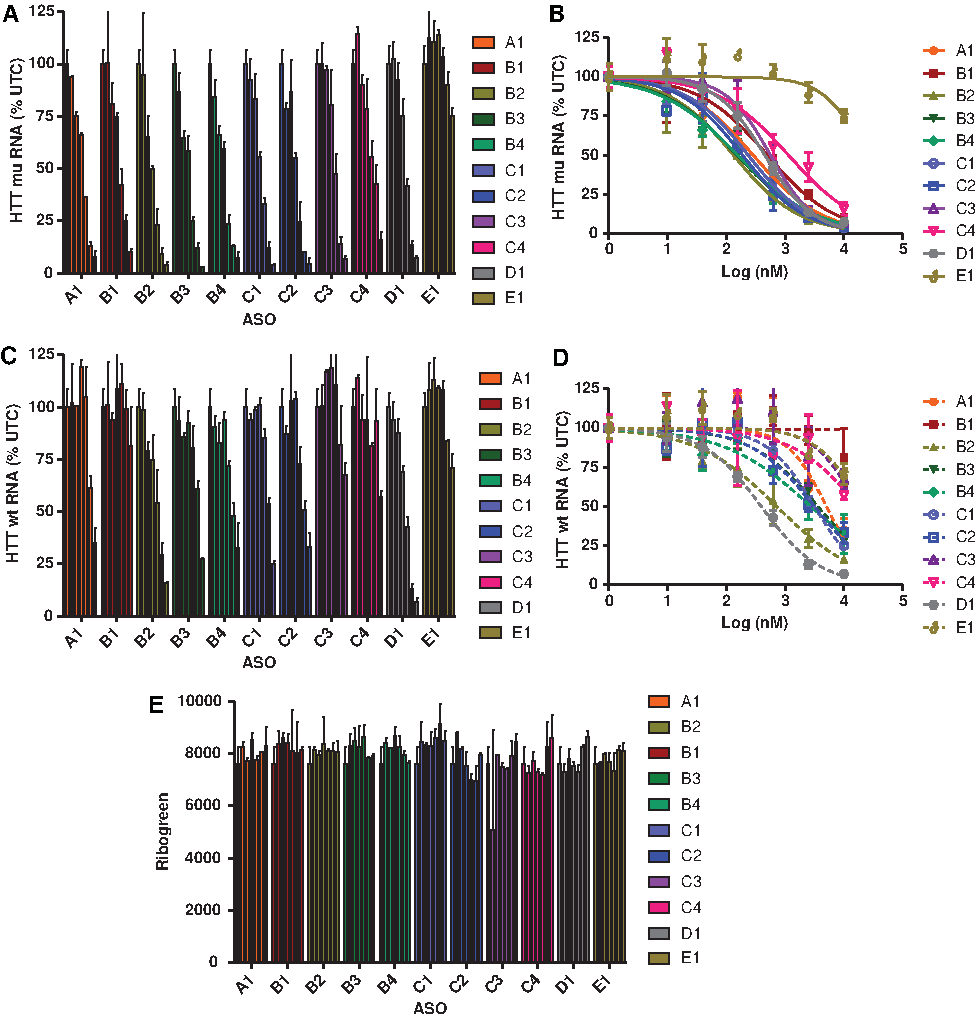
Reduction of
In comparison, introducing 5-TrPh-dT at positions 5, 6, and 11 maintained or reduced potency versus the mutant allele, but did not enhance selectivity compared to parent ASO A1. Interestingly, placing 5-TrPh-dT across from the SNP site (position 8) reduced potency 2-fold versus the mutant allele, but appeared to enhance selectivity versus the wild-type allele relative to parent ASO A1. The slightly higher selectivity observed with ASOs B3 and C3 relative to parent ASO A1 could be attributed to the enhanced binding discrimination for the TG mismatch (Table 2), as a result of introducing the chemical modifications directly across from the SNP site. All ASOs were well tolerated and showed no reductions in total RNA as a measure of nonspecific cellular toxicity (Fig. 2E).
Effect of 2S-dT and 5-TrPh-dT on RNase H cleavage versus wt and mu HTT RNA
We mapped the cleavage sites produced by recombinant human RNase H1 on ASO A1/
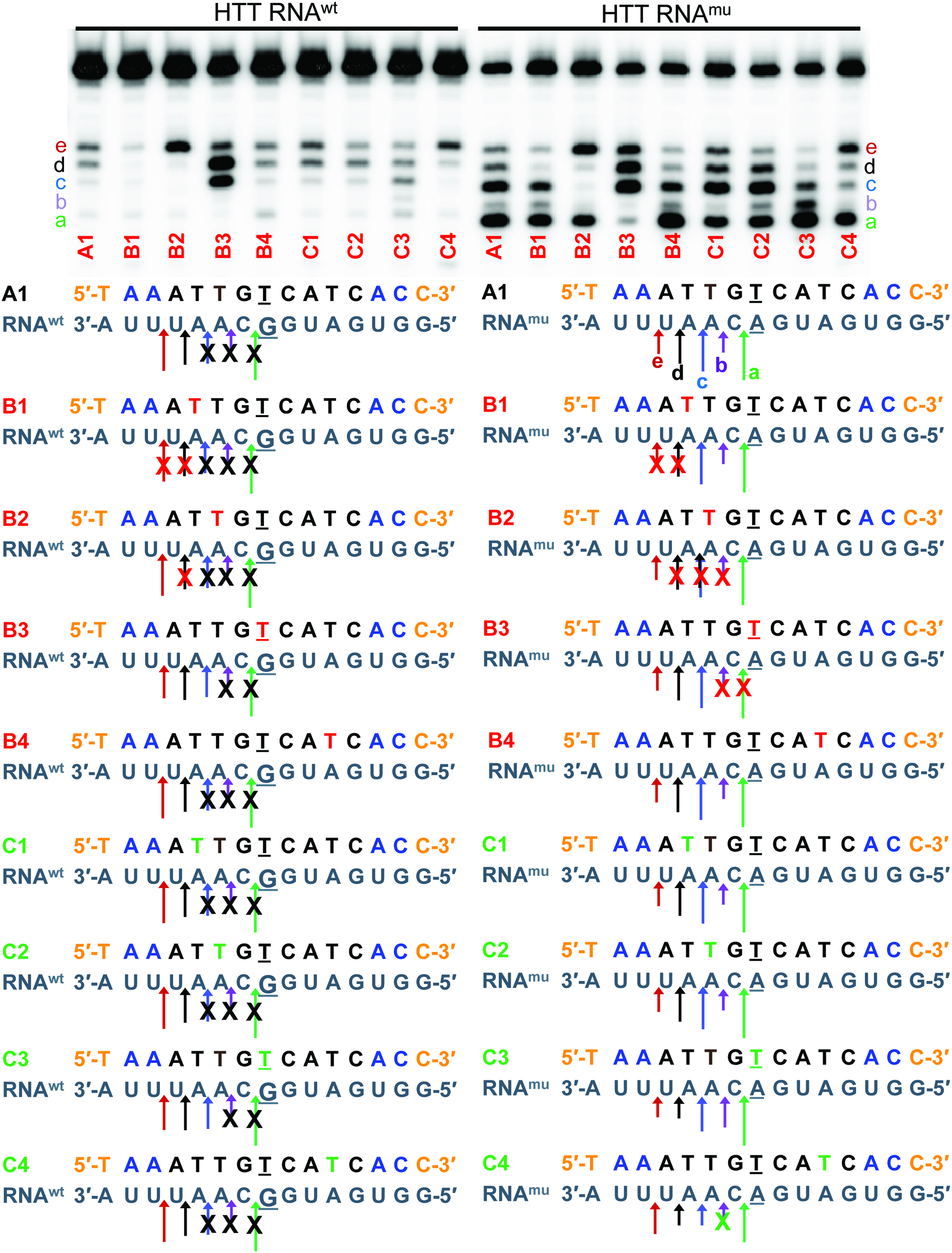
Mapping cleavage sites (a, b, c, d, and e) of recombinant human RNase H1 on ASO/RNA heteroduplexes with matched
Introducing 2S-dT at position 5 of the ASO B1 ablated cleavage sites
We also mapped the RNase H cleavage patterns for 5-TrPh-dT modified ASOs C1-C4 heteroduplexes with the matched
The relationship between magnitude of RNase H1 cleavage of
Structural model for differential effects of 2S-dT and 5-TrPh-dT on RNase H processing
To better understand these observations, we created a structural model of 2S-dT and 5-TrPh-dT modified DNA/RNA duplexes bound to the catalytic domain of human RNase H1 (Fig. 4A). The human enzyme comprises three domains—the catalytic, linking, and hybrid binding—and differs from the Escherichia coli enzyme, which only has the catalytic domain [11,29]. In the context of gapmer ASOs, the hybrid-binding domain is believed to interact with the 3′-wing region [30], while the catalytic domain has a seven-nucleotide footprint on the ASO/RNA heteroduplex (Fig. 4A) [27]. Crystal structures of the catalytic and hybrid domains bound to DNA/RNA hybrids have been reported previously [27,31]. Since human RNaseH1 will have a partially overlapping but distinct seven-nucleotide footprint for every cleavage site, we mapped the interactions of the 2-thio atom and the bulky 5-TrPh group with the enzyme for one cleavage site on the RNA to simplify the analysis (Fig. 4B).
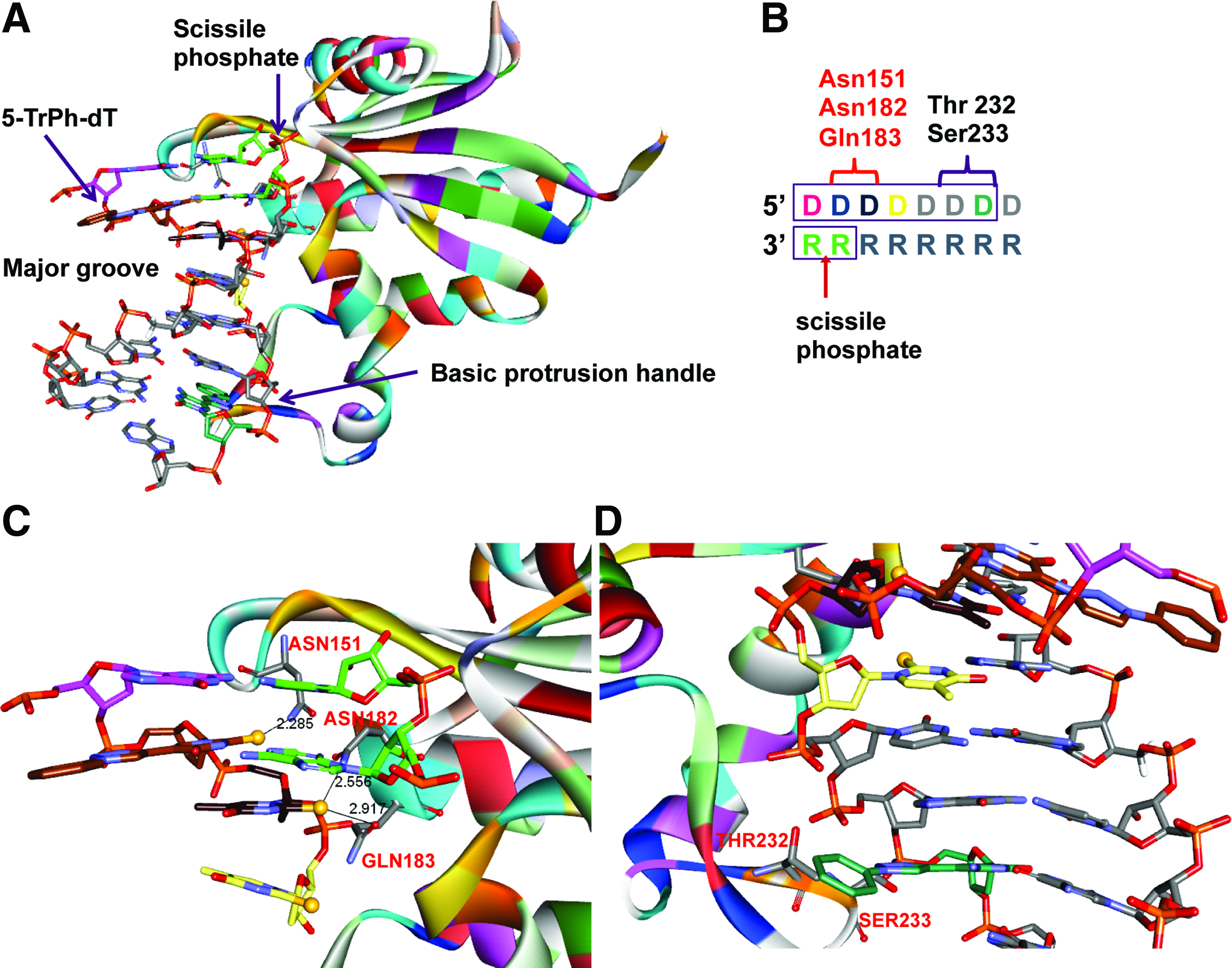
Structural model of the catalytic domain of human RNase H1 bound to a DNA/RNA heteroduplex showing
2S-dT at position 2 (brown carbons) in the DNA strand relative to the scissile phosphate (light green carbons) can result in a tight contact with the side chain of ASN151, while 2S-dT at position 3 (black carbons) can result in a tight contact with ASN182 and GLN183 (Fig. 4B, C). In contrast, 2S-dT at position 4 (light yellow carbons) is not in close proximity to the protein backbone or side chains. Presumably, the tight contacts with the sulfur atom in 2S-dT at positions 3 and 4 are alleviated in case of dT because of the smaller van der Waals radius of oxygen. Superimposition of the model onto the cleavage sites observed in the biochemical assay rationalize loss of cleavage site
In contrast, the bulky 5-TrPh substituent projects into the major groove away from the enzyme, when introduced at position 2 (brown carbons) in the DNA strand relative to the cleavage site on the RNA (Fig. 4B, D). Introducing this substituent at position 7 (olive green carbons) on the DNA strand brings it in close proximity of SER133 and THR232, which are part of the basic protrusion region of the catalytic domain [27]. These observations explain why this modification did not modify the RNase H cleavage patterns when introduced in the proximity of the cleavage sites but appears to modulate RNase H cleavage when incorporated 5–7 nucleotides in the DNA strand away from the cleavage site on the RNA (Fig. 2).
Discussion
HD is an autosomal dominant genetic disorder caused by expansion of a polyglutamine-encoding CAG tract within exon 1 of the HTT gene [1]. Oligonucleotide-based strategies are ideally suited as potential HD therapeutics because of their ability to target the mRNA directly and suppress production of the disease causing protein. Nonallele-selective RNaseH ASOs [32] and siRNA [33], which target homologous sequence tracts in mu HTT and wt HTT mRNA, partially lower both allelic variants and produce therapeutic benefits in murine HD disease models. Allele selectivity can be achieved by targeting the CAG tract directly [34] or by targeting SNPs tightly linked to the CAG expansion [35].
ASOs targeting SNP rs7685686_A in intron 42 of the HTT gene can selectively suppress mutant HTT mRNA and HTT protein in human patient fibroblasts [12,36]. In addition, these ASOs were well tolerated in wild-type rodents and produced potent and sustained suppression of mu HTT protein in the CNS in transgenic murine HD models. However, ASOs targeting SNP rs7685686_A can only provide an allele-selective therapeutic option for ∼45% of the Caucasian HD population due to heterozygosity of SNPs in the broader population. Therefore, additional ASOs at other SNP sites within the HTT gene are required to provide an allele-selective option for all patients.
Recent studies have shown that ASO structure–activity relationships (SAR) for targeting other SNPs can be somewhat unpredictable because of differences in RNase H processing based on changes in the ASO sequence, position of SNP in the gap, and nature of the mismatch [36,37]. Thus, additional chemical strategies, which can improve allele selectivity by enhancing mismatch discrimination or modulating RNase H cleavage of singly mismatched heteroduplexes, are desirable. In this context, our data contribute to the chemical toolbox and provide design options for enhancing properties of SNP targeting ASOs for treating autosomal dominant disorders.
A few previous studies have characterized the effect of non-DNA-like chemical modifications on activity of human RNase H1 in biochemical experiments [38]. However, translating these observations from biochemical systems to real-life examples in cells and animals is lacking. In this broader context, targeting SNP rs7685686_A provides an experimental system to characterize the effects of introducing non-DNA-like chemical modifications within the gap region on the activity and selectivity profile of RNase H ASOs in cells and in animals [12,39,40].
While we were able to provide a rationale for the changes in allele selectivity by mapping RNase H1 cleavage sites on the fully matched versus singly mismatched RNA/ASO heteroduplexes, gaps in our knowledge still remain. For example, the extent of cleavage of
Another confounding variable is that the SNP rs7685686_A is located in intron 42 of the very large HTT pre-mRNA, suggesting that the kinetics of intron splicing could play a role as well. Nonetheless, the most selective ASO in cell culture (B1) produced the slowest (7%) cleavage of
Our structural analysis showed that the sulfur atom in 2S-dT and the 5-triazolylphenyl group in 5-TrPh-dT occupy very distinct sites (Watson–Crick base pairing face versus major groove) in the RNase H1/heteroduplex complex. The C2-position of the pyrimidine nucleobase is in close proximity to highly conserved amino acids ASN 151, ASN182, and GLN 183 in the catalytic domain of RNase H1 [27], when this modification is inserted in the antisense DNA strand within two nucleotides of the cleavage site on the RNA. Replacing the 2-oxygen atom in pyrimidine nucleobases with sulfur causes tight contacts with these amino acids because of the larger van der Waals radius of sulfur causing a shift in the RNase H1 cleavage pattern toward the 3′-side on the RNA strand.
In contrast, the 5-triazolylphenyl group in 5-TrPh-dT is bigger than the sulfur atom in 2S-dT, but its steric bulk projects into the major groove away from the RNase H1-binding interface, when this modification is introduced in the vicinity of the RNase H cleavage site. However, this modification experiences a steric clash with THR232 and SER233 in the basic protrusion handle of human RNase H1 when it is introduced on the DNA strand 5–7 nucleotides away from the cleavage site on the RNA. Despite this steric clash, this modification has a smaller impact on RNase H1 activity, suggesting that the enzyme can accommodate this chemical modification.
In conclusion, we report the synthesis and biological evaluation of gapmer ASOs modified with bulky C5-triazolylphenyl and 2-thio substituted 2′-deoxythymidine nucleotides. We show that single incorporation of these modifications in the gap region does not reduce activity relative to PS DNA and can provide meaningful improvements in allele selectivity. Furthermore, our analysis illuminates the structural basis for the improvements in allele selectivity observed by introducing non-DNA-like chemical modifications in the gap region of ASOs, which function through the RNase H antisense mechanism.
Footnotes
Author Disclosure Statement
No competing financial interests exist. M.E.O., J.N., A.W., and P.P.S. are employees of Isis Pharmaceuticals, Inc.