
Abstract
The discovery of microRNAs (miRNAs) has added a new dimension to the gene regulatory networks, making aberrantly expressed miRNAs as therapeutically important targets. Small molecules that can selectively target and modulate miRNA levels can thus serve as lead structures. Cationic cyclic peptides containing sugar amino acids represent a new class of small molecules that can target miRNA selectively. Upon treatment of these small molecules in breast cancer cell line, we profiled 96 therapeutically important miRNAs associated with cancer and observed that these peptides can selectively target paralogous miRNAs of the same seed family. This selective inhibition is of prime significance in cases when miRNAs of the same family have tissue-specific expression and perform different functions. During these conditions, targeting an entire miRNA family could lead to undesired adverse effects. The selective targeting is attributable to the difference in the three-dimensional structures of precursor miRNAs. Hence, the core structure of these peptides can be used as a scaffold for designing more potent inhibitors of miRNA maturation and hence function.
Introduction
M
miRNAs of the same family can have bivalent roles in cancer. For example, most of the let-7 family members are downregulated in lung cancer [24]. But one member of this family, let-7a-3 has increased expression in human lung adenocarcinoma epithelial cells and has oncogenic function owing to its epigenetic regulation [25]. In certain cases, miRNAs are differentially expressed and show tissue-specific expression [26,27]. Knocking down an entire miRNA family would not be important and significant in such cases. There are other instances in which miRNA of the same seed family have differences in the 3′ end of the sequence, contributing to nonoverlapping targets and hence different functions [28]. In such cases, for understanding the function of one family member and its effect on target, it is necessary to selectively knock down that member, but not the entire family. Hence, selective knockdown is required, but lacking. Our cyclic peptides are one of the scaffolds that provide such selectivity. Polymyxins, a group of cyclic peptides have been recently reported to selectively bind to 18SrRNA, but not 16SrRNA and inhibit eukaryotic translation [29].
Cyclic peptides are peptides wherein the amino and the carboxyl group are linked with a peptide bond. Linear peptides suffer from poor pharmacokinetics, are vulnerable to enzymatic degradation, and the inherently flexible nature allows binding to nonspecific targets [30]. Cyclization of peptides confers several desirable features to be used for targeting RNA. Cyclic peptides, in contrast to linear counterparts have highly ordered structures that confer higher metabolic stability and bioavailability. Conformationally constrained cyclic peptides have minimum binding to nontarget molecules [31,32]. Cyclic peptide mimetics have been used to target TAR RNA interaction with TAT protein essential for HIV viral replication [33–36].
Gramicidin S (GS) is a cyclic amphipathic decapeptide that has been used as an antibiotic against Gram-negative and Gram-positive bacteria. The use of GS as a drug for systemic infections is restricted due to its cytotoxicity and hemolysis of hRBC at low concentrations (μg/mL) [37]. To overcome this hurdle, attempts to design analogs based on GS core structure has been accelerating [38]. In another attempt, recently synthesized derivatives of GS, cyclic peptide-1 (CP-1: C60H94N12O12S22+), cyclic peptide-2 (CP-2: C60H94N12O12S22+, diastereomer of CP-1), and cyclic peptide-3 (CP-3: C60H94N12O12) (Fig. 1) show reduction in cytotoxicities [21]. The primary, but not the only mode of action of GS, is by destabilizing integrity of bacterial membranes. But it has also been reported to interfere with RNA polymerase binding to DNA and inhibiting transcription [39,40]. In addition to this, it has also been used successfully as a transfection agent [41]. The excellent cell-penetrating property and cationic charge on these cyclic peptides allowed us to envisage whether these cationic cyclic peptides can bind to pre-miRNA secondary structure mainly through electrostatic interactions and modulate miRNA levels. Herein, we report three cationic cyclic peptides containing sugar amino acids that can selectively distinguish between the same family members of miRNA.

Chemical structures of sugar amino acid containing cyclic cationic peptides (CP-1, CP-2, and CP-3).
Materials and Methods
Synthesis of cyclic peptides
The cationic cyclic peptides were synthesized as described previously [42].
Cell culture
MCF-7 cells were cultured according to the guidelines provided by ATCC (www.atcc.org) and incubated at 37°C in 5% CO2 incubator. Cells were maintained in Dulbecco's modified Eagle's minimum essential medium (DMEM) High Glucose GlutaMAX™ (Life Technologies) supplemented with heat-inactivated 10% fetal bovine serum, FBS (Life Technologies) and subcultured every other day.
miRNA expression profiling
The MCF-7 cells were seeded in six-well plates (3 × 105 cells/well), grown to 70% confluency, and treated with 10 μM of each of the cyclic peptides (CP-1, CP-2, CP-3). Following treatment, the plates were incubated at 37°C for 48 h in 5% CO2 incubator. The media were removed post 48 h and cells were washed gently with 1× PBS buffer. Total RNA was isolated using TRIzol® Reagent (Invitrogen). Eight hundred nanograms of starting RNA was used for the first-strand cDNA synthesis using universal reverse primer in the Human miRNome Profiler Kit (SBI). MiRNA profiling was done in preformatted quantitative polymerase chain reaction array format as instructed by System Biosciences (SBI). We designed primers for 96 miRNAs, which are well documented for being therapeutically important targets in cancer. Forward primers used for profiling are listed in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/nat).
Real-time quantitative polymerase chain reaction
The MCF-7 cells were passaged in 24-well plates (4 × 104 cells/well), grown to 70% confluency, and treated with a range of different concentrations of the cyclic peptides from 1 to 50 μM. Cells were then incubated at 37°C for 48 h (DMEM, 5% CO2). Following media removal, cells were washed gently with 1× PBS buffer and total RNA was isolated using TRIzol Reagent (Invitrogen). Expressions of mature miRNAs were determined by the QuantiMir Kit (catalog number RA660A-1; SBI). The miRNA expression levels were detected using SYBR Green I PCR Master Mix (Applied Biosystems) on Roche LightCycler LC 480. The relative expression of miRNA was calculated using ΔΔCt method [43] and normalized with respect to U6snRNA as a reference gene. Forward primers were purchased from Sigma (Supplementary Table S1) and the universal reverse primer was provided in the QuantiMir Kit.
In vitro transcription of pre-miRNAs
The pre-miRNAs (sequences shown in Supplementary Table S1) were made from double-stranded DNA template oligonucleotides using the MEGAscript ® T7 High Yield Transcription Kit (Ambion, Inc.). Briefly, linearized ds templates (containing site for T7 RNA polymerase) for in vitro transcription were made by annealing and extending forward and reverse primers (2 μM each) for the respective miRNAs. The forward primer and reverse primers used for constructing templates are given below with the T7 promoter sequence at the 5′ end in bold letters:
For pre-miR-106a:
Forward primer: 5′
Reverse primer: 5′CCATGGTAATGTAAGAAGTGCTTACATTGCAGTAGATCTCAAAAAGCTACCTGCACTGTAAGCACTTTTACATGGCCAAG3′
For pre-miR-106b the primers used were:
Forward primer: 5′
Reverse primer: 5′CCTGCTGGAGCAGCAAGTACCCACAGTGCGGTAGCACGGAGAGGACCACTATCTGCACTGTCAGCACTTTAGCCCCGGCA3′
For pre-miR-30a, the primers were:
Forward primer: 5′
Reverse primer: 5′GCAGCTGCAAACATCCGACTGAAAGCCCATCTGTGGCTTCACAGCTTCCAGTCGAGGATGTTTACA3′
For pre-miR-30b, the primers were:
Forward primer: 5′
Reverse primer:
5′TCCAAGTCAGCTGAAGTAAACATCCACCTCCCAGCCAATCCATGTATTACAGCTGAGTGTAGGATGTTTACATGAACTG3′
The respective primers were extended using Taq polymerase (5 U), dNTPs (0.2 mM), Taq polymerase buffer (1×), and MgCl2 (2 mM). The reaction mixture was denatured by heating at 95°C for 5 min followed by snap chilling on ice for 10 min, followed by primer extension at 72°C for 30 min. The hybrid template with T7 promoter was then used for in vitro transcription following the manufacturer's instructions (Ambion, Inc.). The pre-miRNA substrates were purified by NucAway Spin Columns (Ambion, Inc.) and checked on 15% denaturing polyacrylamide gel electrophoresis.
Dicer blockade assay
The Dicer processing reactions were performed for 4 h at 37°C with the Turbo Dicer (Genlantis) of 1:5 dilutions. Initially, ∼60 pmoles of pre-miRNA (pre-miR-106a, pre-miR-106b, pre-miR-30a, and pre-miR-30b) were labeled with [gamma-32P] ATP using the KinaseMax™ Kit (Ambion). Each pre-miRNA of ∼40,000 cpm was incubated in reaction buffer in a total volume of 8 μL at 37°C. The reaction buffer contained 10 mM sodium cacodylate buffer, 1 mM MgCl2 and 10 mM NaCl (pH 7.5). The reaction mixture was heated at 90°C for 5 min followed by slow cooling at room temperature to 37°C. Different dilutions of Dicer and different concentrations of cyclic peptides were standardized with each pre-miRNA as the relative cleavage efficiency of Dicer varies with different hairpin structures [44]. After standardization, CP-1 (100 nM), CP-2 (3 μM), and CP-3 (1 μM) were added to pre-miR-106a and pre-miR-106b. Similarly, CP-1 (750 nM), CP-2 (1 μM), and CP-3 (750 nM) was added to pre-miR-30a and pre-miR-30b incubated for 30 min at room temperature, followed by the addition of 1 μL of Dicer. As untreated control, same set of pre-miRNAs were incubated with Dicer alone, in the absence of cyclic peptide. After the reaction, an equal volume of 2× sample buffer (95% formamide, 10 mM EDTA, and 0.1% bromophenol blue) was added and loaded on a 15% denaturing polyacrylamide gel. After overnight exposure of the gel to phosphor screen, data were analyzed using a Typhoon PhosphorImager (GE Healthcare). The sequence used for the miRNAs were taken from miRBase, as in the case of other studies [45,46], which consists of mature miRNA sequence and few extra nucleotides upstream. The sequences for precursor miRNAs are listed in Supplementary Table S2. In addition to this, T7 RNA polymerase tends to add few extra nucleotides at the 3′ end during in vitro transcription [14]. Thus, the Dicer cleavage product does not migrate exactly at the size of mature miRNA when electrophoresed in 15% denaturing PAGE.
Results and Discussions
miRNAs are thought to influence pathophysiology of almost all types of cancer. Selective knockdown of overexpressed miRNA is, thus, of sheer importance. Taking cancer as a model system, we screened for expression changes of 96 cancer- related miRNAs (oncomiRs and tumor suppressors) following treatment with each of the three cationic cyclic peptides (CP-1, CP-2, and CP-3) at 10 μM for 48 h in the MCF-7 cell line. These miRNAs were selected based on therapeutic importance as documented and functionally validated in the literature. The list is given in Supplementary Tables S3 and S4. Upon real time quantitative PCR (qPCR)-based profiling of these 96 miRNAs, lowly expressed miRNAs in the MCF-7 cell line were eliminated (18 ≤ ct ≤ 30) and the final heat map of 48 miRNAs was generated (Fig. 2). Upregulated miRNAs are highlighted in dark gray, downregulated miRNAs are highlighted in light gray, and miRNAs whose expression change as compared to the untreated control is not significant are shown in black. We selected two candidate miRNAs (40% downregulation as the cut off), which are commonly downregulated by these three cyclic peptides suggesting the role of GS scaffold to be involved in miRNA modulation.

Heat map summarizing expression profiles of 48 microRNAs (miRNAs). Differential expression of miRNAs upon treatment with each of the cyclic peptide (CP-1, CP-2, and CP-3). Colors range from dark gray (high expression) to light gray (low expression). Black arrows indicate the selected candidate miRNAs that were commonly downregulated by all three cyclic peptides.
The profiling findings of the expression changes in miRNA levels were further validated by qPCR. The MCF-7 cells were treated with each of the three cyclic peptides at a concentration ranging from 1 to 50 μM for 48 h and the expression changes were measured by real time qPCR for the two candidate miRNAs. We observed an inverse correlation between the dose of cyclic peptides and the miRNA levels, that is, decrease in miRNA levels with increase in concentration of each of the cyclic peptides (Fig. 3 and Supplementary Figs. S1 and S2). Upon closer inspection of the profiling data, we found that miR-30a-5p revealed no significant change in its levels following treatment with each peptide, whereas miR-30b was commonly downregulated by all of the three peptides, providing the initial clue of selectivity of these molecules to miR-30b. Similarly, the expression pattern for miR-106a did not significantly change after treatment, whereas miR-106b was commonly downregulated.

Expression validation of miRNA levels by real time qPCR. Concentration-dependent downregulation of
Mature miRNA sequences that differ in few nucleotides are annotated as members of the same family [47]. The differences in the nucleotides in mature forms are reflected in the 3D structures of pre-miR-106a and pre-miR-106b (Supplementary Fig. S4). Processing of precursor into mature form requires catalysis by Dicer. Differences in the pre-miRNA structure, especially in the loop regions or bulges, might affect processing of these precursors [48].
To determine whether these cationic cyclic peptides selectively target pre-miRNA structures, in vitro Dicer cleavage assay was performed. We radiolabeled the 5′ end of pre-miR-106a, pre-miR-106b, pre-miR-30a, and pre-miR-30b. For, pre-miR-106 family pair, we incubated both the miRNAs in the presence and absence of CP-1, CP-2, and CP-3. Following incubation with each molecule, Dicer was added and reaction was allowed to proceed. Blocking of processing by cyclic peptides was observed only in case of pre-miR106b (Fig. 4), whereas band intensity in case of pre-miR-106a in the presence and absence of each cyclic peptide remained almost unchanged. We speculated that there must be differences in secondary structures of pre-miRNAs even though within the same family. Using Mfold server, we determined the putative secondary structures of pre-miRNAs and found that despite the mature sequence of a miRNA family being highly similar, there is difference in the precursor stem loop structure. There is an extra bulge in the stem region in pre-miR-106b, which might be amenable to cyclic peptide binding and hence preventing Dicer processing. However, pre-miR-106a lacks these extra secondary structures making it less likely for recognition and binding of these cyclic peptides (Supplementary Fig. S4). Similar results were obtained for miR-30 family members, whereby processing of pre-miR-30b was selectively impaired by each of the cyclic peptides, but not for pre-miR-30a (Supplementary Fig. S3). These results correlate with the structural differences between pre-miR-30a and pre-miR-30b (Supplementary Fig. S5).
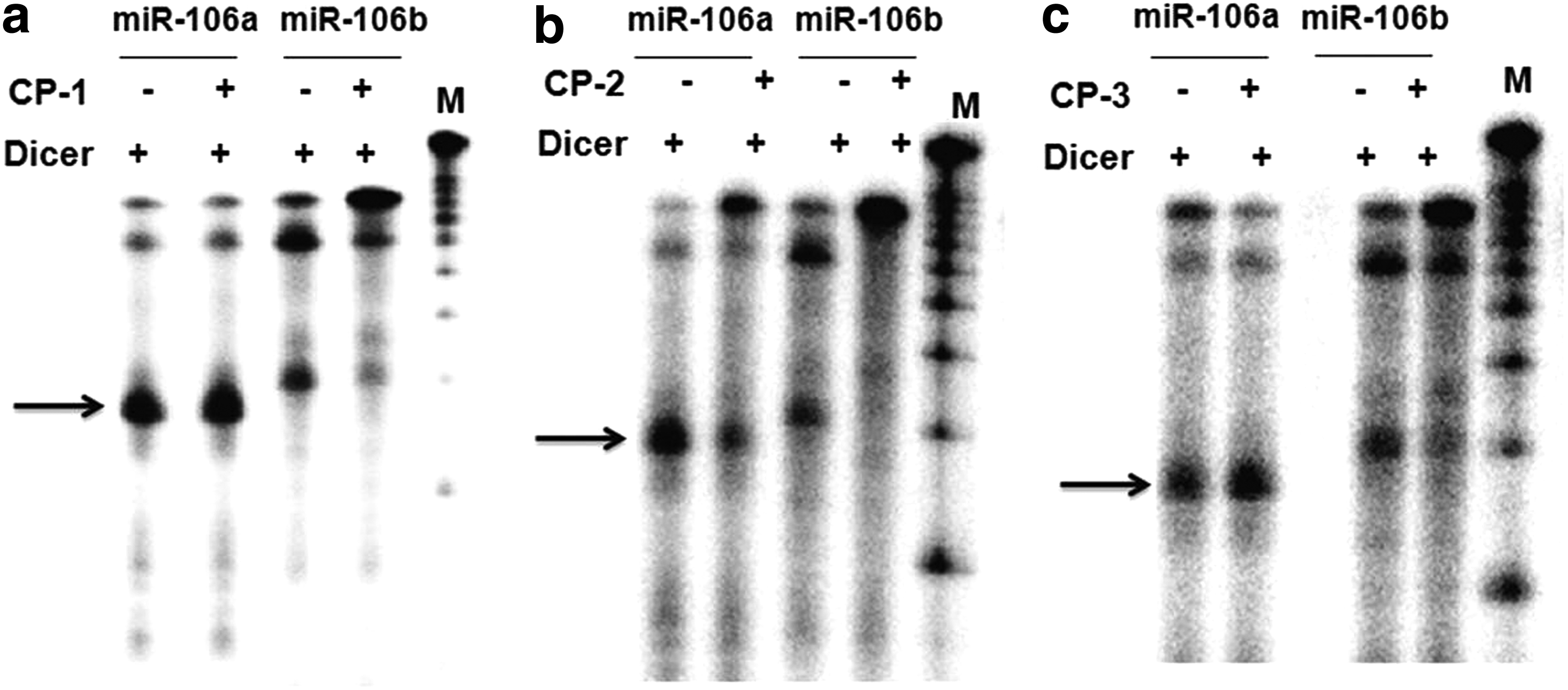
Dicer blockade assay. In vitro synthesized and 5′ end radiolabeled pre-miR-106a and miR-106b untreated and treated with
Conclusions
In summary, the results presented here highlight the ability of three cationic cyclic peptides to downregulate selective family members of miRNA by hindering its maturation process. The promising area of miRNA as a target for pharmaceutical intervention requires identification of biologically active, low-molecular weight, cell-permeable molecules that can target secondary structure of RNA selectively. miRNAs within the same family might have bivalent roles or tissue-specific expression. In such cases, ability to selectively distinguish and knockdown of one miRNA family member is imperative, but lacking. Cationic cyclic peptides provide a new class of scaffolds that can target pre-miRNA structures selectively. Lead optimization of this scaffold can further augment the selectivity for its target, thus paving the way for its use in therapeutics.
Footnotes
Acknowledgment
This work was supported by the Council of Scientific and Industrial Research (CSIR) (Project title: Genome Dynamics in Cellular Organization, Differentiation, and Enantiostasis Project code BSC0123), India.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.