
Abstract
The Hsp70 chaperone system plays an important role in protein quality control by assisting in the folding and clearance of misfolded proteins. However, the mechanism by which it chooses between folding and degradation pathways is not fully understood. In this study, we used an RNA aptamer for Hsp70 to perturb the function of Hsp70 in cell-free systems. We found that the aptamer inhibited both Hsp70-mediated folding and Hsp70-CHIP-mediated ubiquitination/degradation of a misfolded protein substrate. Based on these results, we explored a novel strategy for targeted protein ubiquitination, using an engineered bifunctional aptamer to tether a protein substrate to Hsp70. We demonstrated that increased Hsp70-CHIP-mediated ubiquitination of the tethered protein substrate can be specifically induced by this bifunctional aptamer. This strategy may be useful in selective degradation of disease-causing proteins for therapeutic purposes. In addition, these studies provide insight into the mechanism of Hsp70-mediated protein triage.
Introduction
I
Hsp70 has two domains, a 45 kDa N-terminal nucleotide binding domain (NBD) and a 30 kDa substrate binding domain (SBD) with a C-terminal lid. ATP is bound and hydrolyzed in the NBD, and the SBD binds to exposed hydrophobic peptide segments of a substrate protein. The conformation of Hsp70 changes extensively between its ATP and ADP-bound states [11–13]. In the ATP-bound state, the lid of the SBD is in an open conformation, resulting in low affinity for substrates [11,13,14]. In the ADP-bound state, the lid is in a closed conformation, resulting in a higher affinity for substrates [12,15]. Hsp70 is equipped with a bidirectional allosteric regulation, in which ATP hydrolysis at the NBD stabilizes substrate interaction at the SBD and substrate binding at the SBD stimulates ATP hydrolysis at the NBD [16]. In this way, Hsp70 continuously binds and releases its substrate accompanied by its ATPase cycle. Unable to act alone, Hsp70 invariably requires a J protein (Hsp40) and sometimes a nucleotide exchange factor as partners [17]. These cochaperones modulate Hsp70 function by regulating its ATPase activity. Hsp40s form a class of diverse proteins, all of which have a conserved J domain. Interaction of the J domain with Hsp70 stimulates the low intrinsic ATPase activity of Hsp70 and accelerates its substrate binding and release cycle [18,19]. Distinct sets of cochaperones also dictate the fate of Hsp70 substrates, whether they will be folded or degraded [20]. For example, CHIP is an E3 ubiquitin ligase, which ubiquitinates Hsp70-bound protein substrates, thereby labeling them for proteasomal degradation [10,21]. CHIP has an N-terminal tetratricopeptide repeat domain that interacts with the C-terminal EEVD motif of Hsp70 and a C-terminal U-box domain, which interacts with ubiquitin-conjugating E2 enzymes [22].
Hsp70 has been shown to be involved in both folding and degradation of a protein [23], which indicates an important role in the choice between folding or degradation pathways for misfolded proteins. The triage decision to degrade chaperone substrates is a tricky balancing act, but one that is critical for efficient quality control, and should be strictly regulated. Extensively misfolded protein substrates should be directed toward degradation, while repairable substrates should be spared to allow refolding. Chaperone substrate degradation is a thermodynamically unfavorable process because both ubiquitination and proteasomal degradation consume ATP, and de novo protein synthesis to replace degraded proteins is also energy intensive [5]. On the other hand, extensively misfolded substrates must be degraded to avoid futile chaperone cycling with them, which also consumes ATP, and to prevent aggregate formation, which is toxic to the cell. Although the identity of Hsp70 cochaperones that enable folding or degradation of its substrates is known [2], the mechanism of pathway selection and regulation is not clear. Hsp70 ATPase activity is required for substrate folding [18,19]. However, the cochaperone CHIP, which promotes substrate degradation, also inhibits Hsp40-stimulated ATPase activity of Hsp70, decreases Hsp70-substrate interaction, and suppresses substrate folding [10,24]. The significance of these CHIP-mediated effects on Hsp70 for the triage process is not clear.
To study the function of Hsp70s and their cochaperones, a combination of genetic and pharmacologic manipulations is often employed. Different factors may be overexpressed in cells [9]; their expression levels may also be reduced using RNAi [25]. However, Hsp70 expression is regulated by a robust heat shock response circuit that modulates cellular Hsp70 levels in response to various stress insults [7], which makes studying individual pathways regulated by Hsp70 in living cells difficult [1]. Given the importance of the ATPase cycle in Hsp70 function, reagents that bind Hsp70-ATP and modulate its ATPase activity directly would be useful in dissecting the protein triage mechanism. For this purpose, we recently generated an Hsp70-ATP conformation-specific RNA aptamer, AptHsp70-1 [26]. Interestingly, the aptamer inhibited both intrinsic and Hsp40-stimulated ATPase activities of Hsp70 [26]. In this study, we utilize this aptamer as a tool in reconstituted in vitro folding and degradation assays to gain insights into the mechanism of this triage process and to explore new therapeutic possibilities. We report that this aptamer destabilized Hsp70-substrate interaction and reduced both Hsp70-mediated folding and Hsp70-CHIP-mediated ubiquitination of a misfolded protein substrate. To implement a strategy for targeted ubiquitination, we engineered a bifunctional aptamer to tether the estrogen receptor α (ERα) to Hsp70. By means of this manipulation, which is not feasible with conventional genetic or pharmacologic methods, we were able to specifically increase ubiquitination of ERα. Based on these results, we also propose a control mechanism for Hsp70-mediated protein triage.
Materials and Methods
Proteins, plasmids, and chemicals
The expression plasmid for human heat shock protein 70 kDa 1A (hHsp70A1A) with a C-terminal His-tag was purchased from Genecopoeia (Cat. No. EX-Z5704-B31). The protein was expressed in Escherichia coli strain BL21-AI and purified by a nickel affinity column (Sigma) according to the manufacturer's instructions. Purified protein fractions were pooled together and concentrated using protein concentrators (9 kDa molecular weight cutoff; Pierce Scientific) and buffer exchanged using the same concentrators into protein storage buffer (50 mM HEPES/pH 7.4, 100 mM NaCl, 3 mM DTT, 2 mM MgCl2, 45% glycerol, 100 μM ATP).
E. coli Hsp40 (DnaJ) was purchased from Enzo Life Sciences. CHIP was purchased from Millipore. ERα was purchased from Invitrogen. UBE1, UbcH5b, ubiquitin, purified 26S proteasome, 10× ubiquitin conjugation reaction buffer, and Mg-ATP were purchased from Boston Biochem.
Aptamers and their derivatives
The sequences of various aptamers used are as follows:
AptHsp70-1: 5′GGGAGAAUUCAACUGCCAUCUAGGCCUUAUAAACAGCCGGAUCCCGAUUGUGCUCGAUAUGUACUCGCACCUUAACUACAAGCUUCUGGACUCGGU3′
AptHsp70-1-66S: 5′GGGCCGAGAAUUCAACUGCCAUCUAGGCCUUAUAAACAGCCGGAUCCCGAUUGUGCUCGAUAUGUACUCGGCCC3′
AptER-1: 5′GGGCAGAGGCACCGCGAACAAAACGCAAGACAGAGUGCCGACAAGAGCACUACAAGCUUCUGCCC3′
AptHsp70-ER-1: 5′GGGCCGAGAAUUCAACUGCCAUCUAGGCCUUAUAAACAGCCGGAUCCCGAUUGUGCUCGAUAUGUACUCGGCCCGCUCGGCACCGCGAACAAAACGCAAGACAGAGUGCCGACAAGAGCACUACAAGCCGAGC3′
Secondary structures were predicted using the Vienna RNA server [27].
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed as described previously [26]. Briefly, 30 nM of 32P-labeled RNA aptamer was incubated with protein in 15 μL binding reactions. The reactions were incubated at 37°C for 40 min, and run on 4.8% polyacrylamide gel (50:1 Acrylamide:Bis for AptHsp70-ER-1 and 37.5:1 Acrylamide:Bis for AptHsp70-1 and AptER-1) in 0.5× TBE buffer. The gels were dried, exposed to phosphor screen, and imaged by Typhoon phosphorimager (GE Healthcare).
Coimmunoprecipitation
Luciferase (0.3 μM) was heat-denatured at 43°C for 5 min with Hsp70 (0.3 μM) in buffer (20 mM HEPES, pH 7.4, 100 mM KCl, 10 mM MgCl2), in the presence or absence of AptHsp70-1, incubated at 4°C for 60 min, then centrifuged at 16,200 g for 10 min at 4°C. To the supernatant, goat anti-human Hsp70 antibody (K-20, Cat. No. sc-1060; Santa Cruz Biotechnology) was added and incubated at 4°C for 60 min, then Protein G PLUS agarose beads (Santa Cruz Biotechnology) were added and incubated at 4°C for 120 min, washed four times with the buffer, and immunoblotted with goat antirecombinant firefly luciferase antibody (Cat. No. G745A; Promega).
In vitro heat-denatured luciferase refolding assay
Luciferase refolding assay was performed as previously described [28,29] with minor modifications. Briefly, 80 nM luciferase, 160 nM DnaJ, and 400 nM Hsp70 (1:2:5) in buffer (25 mM HEPES/pH 7.4, 50 mM KCl, 3 mM MgCl2, 2 mM DTT) with 2 mM Mg-ATP were heat-denatured at 43°C for 10 min in the presence or absence of AptHsp70-1 (Hsp70:Aptamer = 1:4). The mixture was incubated at room temperature for various time periods ranging from 1 to 150 min. Luciferase activity was measured as luminescence using luciferase assay reagent (Promega) according to the manufacturer's instructions.
In vitro ubiquitination and proteasomal degradation assay
In vitro ubiquitination of heat-denatured luciferase was performed as described previously [30] with minor modifications. Hsp70 (0.3 μM), DnaJ (0.3 μM), and luciferase (0.3 μM) in 1× ubiquitin-conjugating buffer (Boston Biochem) with 50 mM KCl, 2.5 mM DTT, and 1 mM Mg-ATP were treated at 43°C for 5 min in the presence or absence of aptamer (Hsp70:Aptamer = 1:5), cooled on ice for 1 min. Ubiquitination mix (0.1 μM UBE1, 2 μM UbcH5b, 1.3 μM CHIP, 150 μM ubiquitin, and 1 mM Mg-ATP) was added, and the mixture was incubated at 30°C. The reaction was stopped at various times as indicated by adding 5× SDS gel loading buffer. Samples were run on 10% SDS-PAGE, transferred onto PVDF membranes, and immunoblotted with goat antirecombinant firefly luciferase antibody (Cat. No G745A; Promega). For proteasomal degradation assay, 30 nM purified 26S proteasome was added to the ubiquitination mix and incubated at 37°C. In vitro ubiquitination of ERα was performed using the same procedure described above, except that there was no heat-denaturing step and immunoblotting was with the rabbit anti-human ERα antibody (HC-20, Cat. No. sc-543; Santa Cruz Biotechnology). The ubiquitinated bands were quantified by Quantity One (Biorad) image analysis software.
Results
AptHsp70-1 destabilizes Hsp70-substrate interaction and reduces Hsp70-mediated folding of a misfolded protein substrate
The ATPase cycle of Hsp70 is coupled to its substrate binding and release. Hydrolysis of ATP would trigger the closing of the substrate binding cavity and the locking-in of the associated substrate. Based on this information, we investigated Hsp70-substrate interaction under aptamer perturbation. For this purpose, we employed a widely used model of misfolded protein substrate for Hsp70, the heat-denatured luciferase [24,29]. In a coimmunoprecipitation assay, luciferase was heat-denatured at 43°C in the presence of Hsp70 and DnaJ, Hsp70 was immunoprecipitated by Hsp70 antibody, and coprecipitated luciferase was detected by western blot. In the presence of the aptamer, ∼40% less luciferase coimmunoprecipitated with Hsp70 (Fig. 1A), which indicated that the aptamer destabilized Hsp70-substrate interaction. Since the aptamer inhibits Hsp70 ATPase activity [26], it may be destabilizing Hsp70-substrate interaction by impeding the transition to Hsp70-ADP state that has a higher affinity for substrates [14,15,31,32]. In addition, the aptamer binding may also sterically block substrate binding to Hsp70. Previously, we demonstrated that the aptamer inhibited Hsp70 ATPase activity in the presence of Hsp40, but did not affect the stimulatory effect of Hsp40 on this ATPase activity [26], therefore the reduction of folding efficiency observed here is unlikely due to a destabilization of interaction between Hsp70 and Hsp40 by the aptamer.
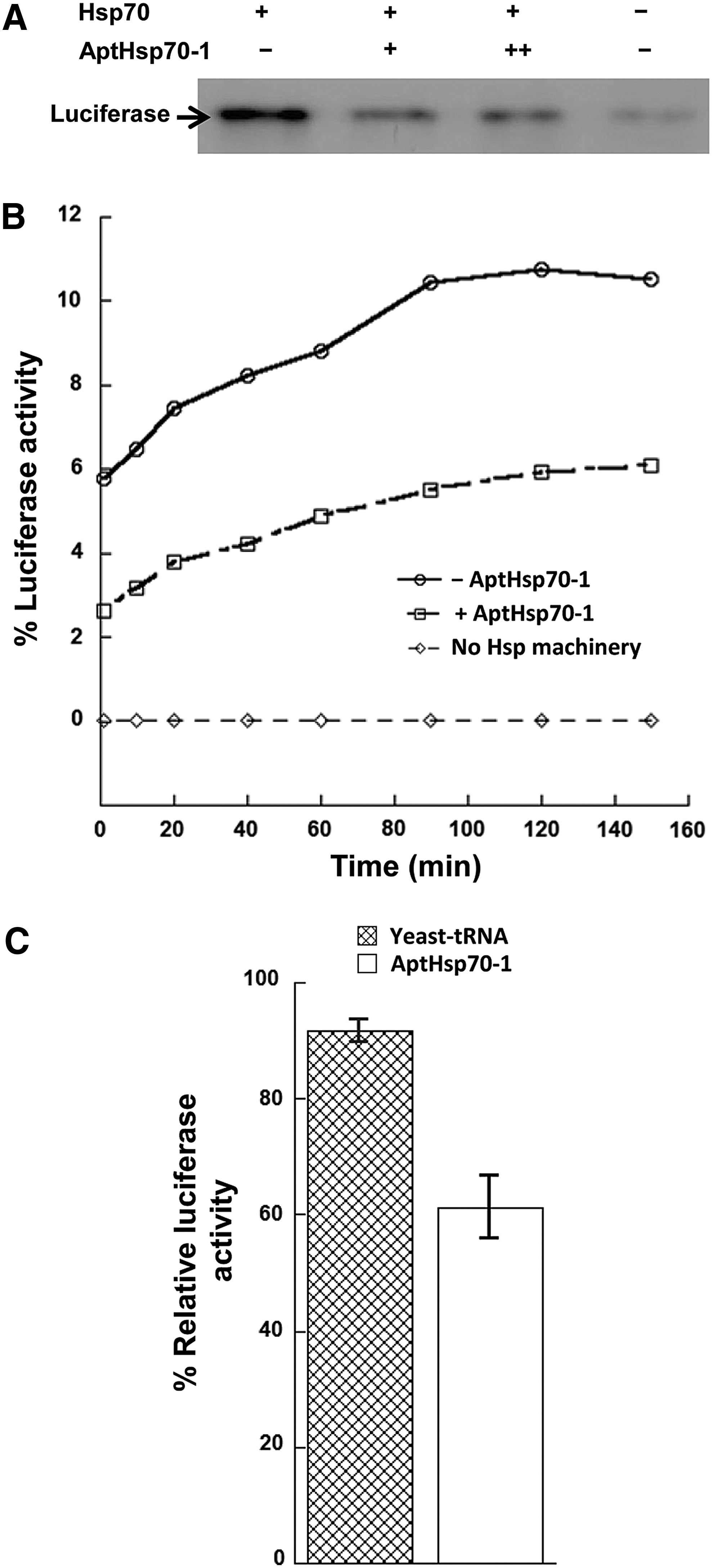
Destabilization of Hsp70-substrate interaction and reduction of Hsp70-mediated folding of denatured luciferase by AptHsp70-1.
The destabilization of Hsp70-subsrate interaction may further affect Hsp70 capability to assist in substrate folding. To study the effect of the aptamer on the folding activity of Hsp70, we used the well-characterized, in vitro reconstituted, denatured luciferase refolding assay [28,29]. Luciferase was heat-denatured in the presence of Hsp70 and DnaJ by heating to 43°C. The denatured luciferase was allowed to refold at room temperature and luciferase activity was measured by luminescence at different time points. As shown in Fig. 1B and C, the aptamer significantly decreased refolding of heat-denatured luciferase compared with nonspecific yeast tRNA. Thus, the aptamer caused suppression of the folding pathway.
AptHsp70-1 reduces Hsp70-CHIP-mediated ubiquitination of a misfolded protein substrate
The E3 ubiquitin ligase CHIP binds to the C-terminus of Hsp70 and connects the Hsp70 chaperone system to the ubiquitination and proteasomal degradation pathway. To study the effect of the aptamer on Hsp70-CHIP-mediated ubiquitination of misfolded protein substrates, we used the in vitro reconstituted ubiquitination assay. In this assay, luciferase was heat-denatured in the presence of Hsp70 and DnaJ at 43°C for 5 min and cooled on ice. CHIP, the E2 enzyme UbcH5b, UbE1 enzyme, and ubiquitin were then added and incubated at 30°C for ubiquitination [30]. As shown in Fig. 2A, when the aptamer was added with Hsp70 and DnaJ during the heat denaturation step, ubiquitination of luciferase was inhibited. Because nondenatured luciferase is not an Hsp70 substrate [21,30], we tested as a control the effect of the aptamer on ubiquitination of luciferase without heat denaturation. As shown in Fig. 2B, the aptamer had no effect on ubiquitination of nondenatured luciferase in the presence of Hsp70 and DnaJ. Taken together, these experiments showed that the aptamer reduced ubiquitination of denatured luciferase by inhibiting Hsp70 activity, not by inhibiting enzymes involved in ubiquitination. To extend this result, we performed a degradation assay, in which purified 26S proteasome was additionally included in the ubiquitination reaction. As shown in Fig. 2C, the aptamer reduced proteasomal degradation of heat-denatured luciferase in this assay, which is consistent with the reduction of Hsp70-CHIP-mediated ubiquitination by the aptamer.
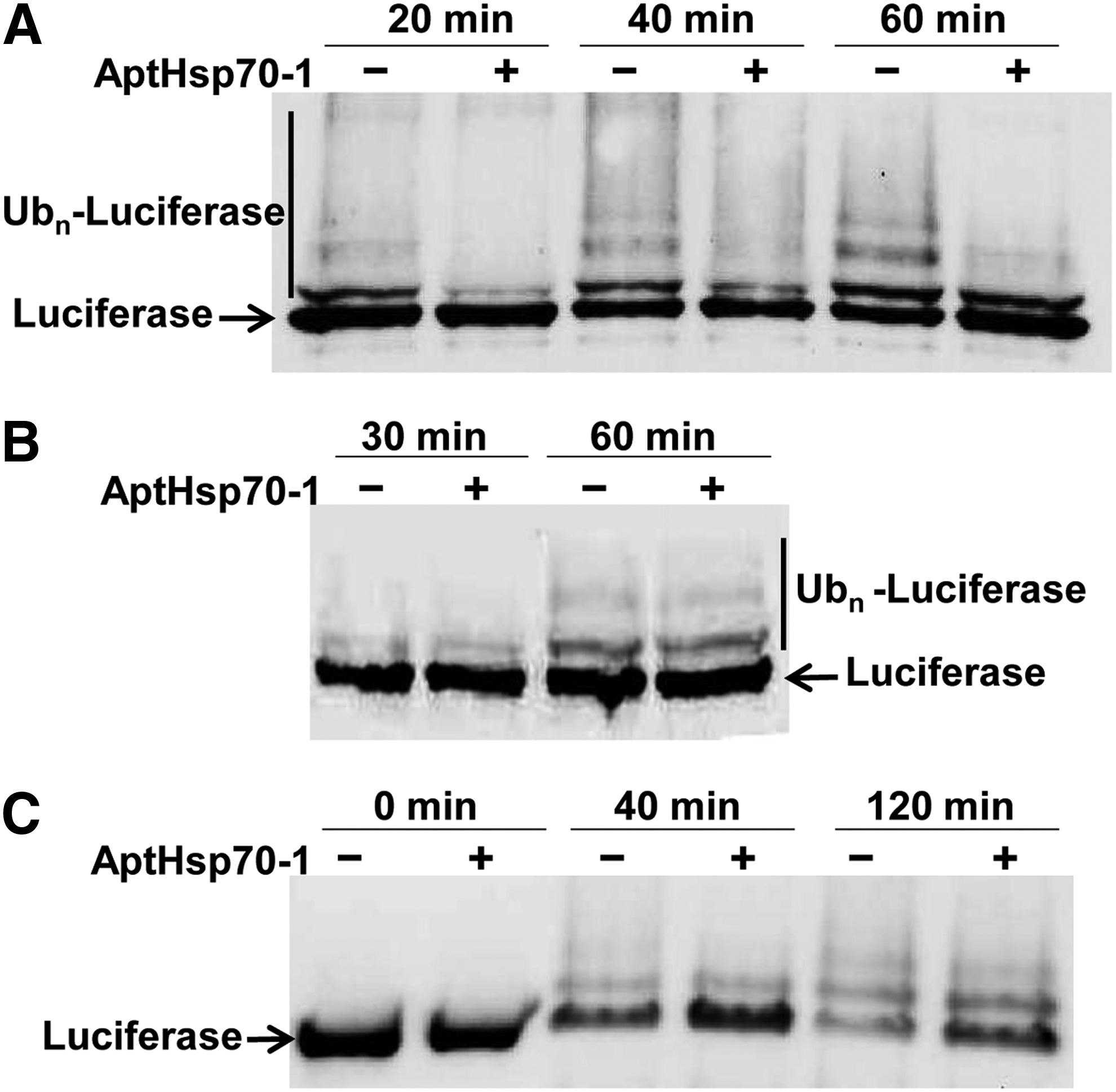
Reduction of Hsp70-mediated ubiquitination of denatured luciferase by AptHsp70-1.
The results presented so far suggest a correlation between the strength of Hsp70-substrate interaction and the extent of Hsp70-CHIP-mediated ubiquitination of the substrate as the aptamer destabilized Hsp70-substrate interaction (Fig. 1A) and also reduced Hsp70-CHIP-mediated ubiquitination of a misfolded protein substrate (Fig. 2A). If there were indeed such a correlation, then conversely, tethering a specific protein to Hsp70 would lead to its increased ubiquitination in the presence of CHIP. Confirming this hypothesis would suggest a novel approach to intentional and selective ubiquitination that might have therapeutic utility. Therefore, we proceeded to create such an aptamer-enabled scenario.
A bifunctional aptamer is created to connect Hsp70 with ERα
By connecting AptHsp70-1 to an aptamer for a substrate protein, we should be able to create a bifunctional aptamer that tethers a specific substrate to Hsp70. However, it may not be possible to generate aptamers to specifically recognize a misfolded protein substrate such as denatured luciferase. So, we turned to a native or near-native Hsp70 substrate for this study. Different from a misfolded protein substrate such as heat-denatured luciferase, this type of Hsp70 substrate has an almost native fold except for certain unstructured regions, which cycle with chaperones to become stabilized [33].
One of the native/near-native Hsp70 substrates is the ERα, which is a nuclear receptor of steroid hormone. Ubiquitination and degradation of ligand-free ERα have been shown to be mediated by the Hsp70-CHIP system [34,35]. Previously, our laboratory had generated and characterized an RNA aptamer to ERα, AptER-1 [36], which has a dissociation constant of 16 nM and could be used to form a bifunctional construct with AptHsp70-1. To ensure the concordance of results between experiments with either denatured luciferase or ERα, we first tested the effect of AptHsp70-1 on Hsp70-CHIP-mediated ubiquitination of ERα by the in vitro ubiquitination assay. Native ERα without heat denaturation was used in the assay since ERα is a native Hsp70 substrate. As shown in Fig. 3A and B with both gel images and bar graphs, we found that AptHsp70-1 reduced Hsp70-CHIP-mediated ubiquitination of ERα, while nonspecific yeast tRNA had no effect on ERα ubiquitination. Therefore, both native ERα and denatured luciferase behaved similarly in this assay.

Engineering an Hsp70-ERα bifunctional aptamer, AptHsp70-ER-1.
We then created the Hsp70-ERα bifunctional aptamer (AptHsp70-ER-1) using the information obtained from characterization of the two aptamers. The minimized Hsp70 aptamer (AptHsp70-1-66S) was connected to the previously defined ERα aptamer [the minimized AptER-1(65nt)] [26,36]. The free stems of the two aptamers that were not involved in binding were linked covalently in one strand to create a single molecular entity (Fig. 3C). The bifunctional AptHsp70-ER-1 was tested for binding to its two protein targets by EMSAs. Whereas the monofunctional AptHsp70-1 did not bind ERα (Fig. 3E) and the monofunctional AptER-1 did not bind Hsp70 (Fig. 3F), the bifunctional AptHsp70-ER-1 was able to bind to both Hsp70 and ERα (Fig. 3D). In the presence of both Hsp70 and ERα, a supershift of the AptHsp70-ER-1 RNA band was observed in EMSA (Fig. 3D), while no supershift of AptHsp70-1 RNA band was observed under the same condition (Fig. 3E). These results indicate that the supershift of AptHsp70-ER-1 was most likely due to simultaneous binding of the aptamer to both Hsp70 and ERα. Thus, there was no steric hindrance to prevent the triple complex formation and the bifunctional construct was capable of tethering ERα to Hsp70.
AptHsp70-ER-1 bifunctional aptamer specifically increases Hsp70-CHIP-mediated ubiquitination of ERα
Next, we used the in vitro ubiquitination assay to study the effect of the bifunctional aptamer on CHIP-mediated ubiquitination of ERα. As shown in Fig. 4A with both gel image and bar graphs, AptHsp70-1 inhibited Hsp70-CHIP-mediated ubiquitination of ERα, while the bifunctional aptamer significantly increased the population of multiubiquitinated ERα (Ubn-ERα) over the level observed when no aptamer was present (compare lanes 1 and 4 with lanes 3 and 6, respectively, in Fig. 4A). Since ERα with one and two ubiquitins attached (Ub1-ERα and Ub2-ERα) becomes multiubiquitinated (Ubn-ERα), the intensity of the bands representing ERα with one and two ubiquitins attached (Ub1-ERα + Ub2-ERα) is lesser in the bifunctional aptamer lanes compared with no aptamer and the monofunctional aptamer lanes (Fig. 4A). Importantly, the bifunctional aptamer remained inhibitory to Hsp70-CHIP-mediated ubiquitination of a nontethered substrate, namely the heat-denatured luciferase (Fig. 4B). This inhibitory effect should have also been present for the nontethered ERα to a degree similar to that revealed by the monofunctional AptHsp70-1 in lanes 2 and 5 of Fig. 4A. Although it is not practical to dissect the kinetics of this multicomponent complex using the current experimental system, increased CHIP-mediated ubiquitination of ERα tethered to Hsp70 through AptHsp70-ER-1 suggests that substrate ubiquitination may not require strict geometric arrangement of the substrate with respect to Hsp70 and CHIP. This is consistent with a previous study, which suggested that CHIP forms a dynamic complex with Hsp70 to search a large space for ubiquitinating Hsp70 substrates [37].

Enhancement of Hsp70-CHIP-mediated ubiquitination of ERα by the AptHsp70-ER-1 bifunctional aptamer.
Discussion
The Hsp70 system acts at a critical juncture of decision making to coordinate both folding and degradation pathways for its substrate proteins. Through aptamer-enabled perturbation of this process, we studied the relationship between the strength of Hsp70-substrate interaction and the extent of Hsp70-CHIP-mediated ubiquitination of the substrate. Previously, we demonstrated the inhibitory effect of AptHsp70-1 on Hsp70 ATPase activity. As both folding and ubiquitination require the ATPase activity, which enables cycling of the substrate with Hsp70, it is not surprising to observe that both folding of protein and its ubiquitination are inhibited by the aptamer. The interesting converse results obtained with the monofunctional AptHsp70-1 and the bifunctional AptHsp70-ER-1 are summarized in Fig. 5A. In particular, the bifunctional aptamer specifically increased the ubiquitination of the tethered ERα, suggesting a novel strategy for targeted ubiquitination. Targeted protein ubiquitination could lead to degradation of intentionally designated proteins, which has potential for biotechnological or therapeutic applications [38,39]. Previously, the Skp1-Cullin-F box (SCF) ubiquitination system was used for targeted protein ubiquitination using bifunctional molecules called protacs (proteolysis targeting chimeric molecules) [38,39]. Similar to the SCF system, our work showed that the Hsp70-CHIP system can also be used for targeted protein ubiquitination. Since Hsp70 is involved in protein quality surveillance in every compartment of the cell, tethering specific proteins to Hsp70 could be an efficient strategy for targeted protein ubiquitination and degradation.
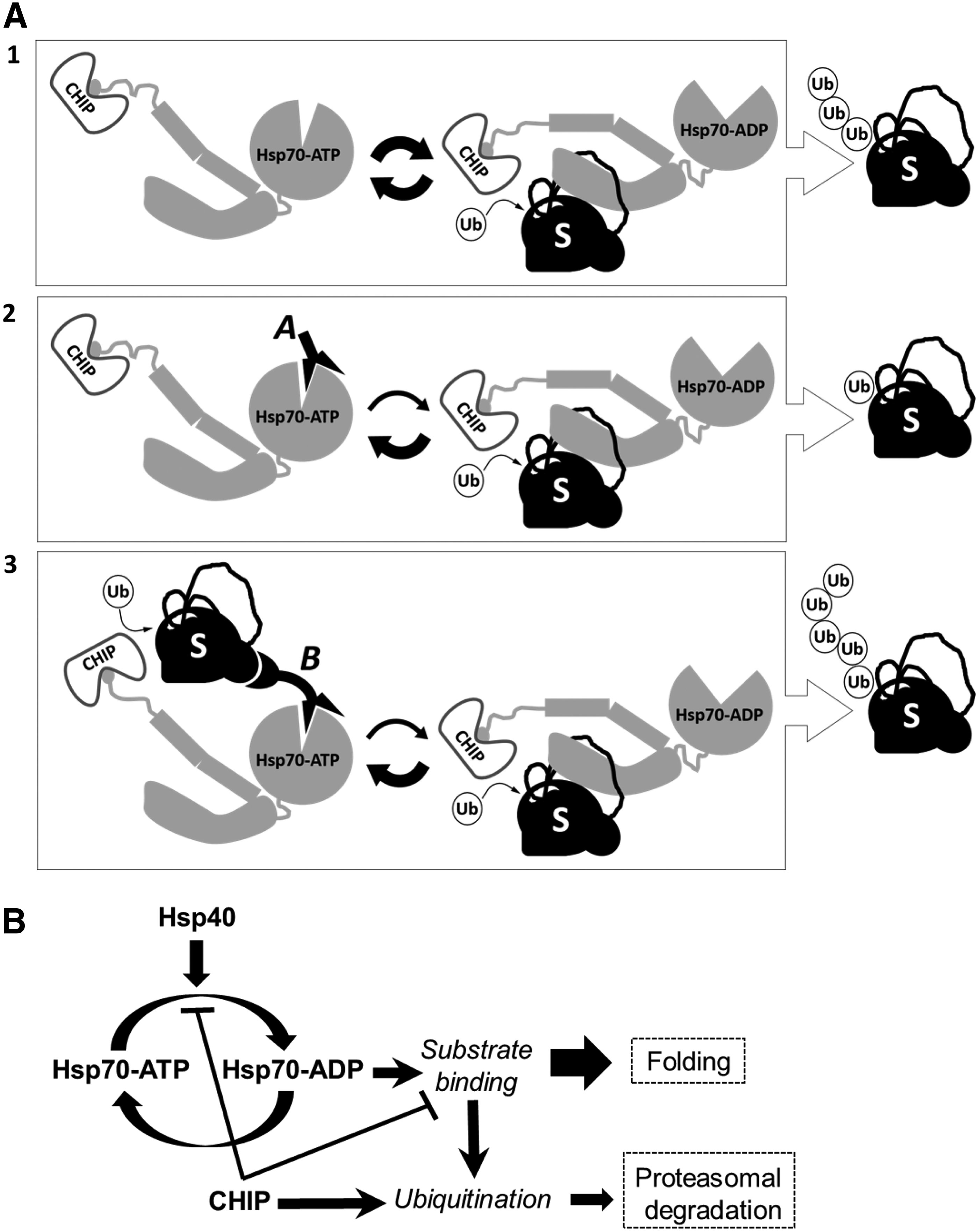
Aptamer perturbation of Hsp70-mediated misfolded protein triage.
In cancer, a higher level of Hsp70 is often correlated with higher tumor grades and poorer survival [40–42]; inhibition of Hsp70 with antisense reagents caused apoptosis of tumor, but not normal cells [25]. In breast cancer, ERα is an important therapeutic target for ER-positive tumors [43,44], and selective ER degradation could be an effective treatment even after they develop resistance to ER antagonists [45,46]. The Hsp70-ERα bifunctional aptamer specifically increased Hsp70-CHIP-mediated ubiquitination of the tethered ERα while inhibiting ubiquitination of a nontethered Hsp70 substrate. This shows that it may be possible to use molecules such as the bifunctional aptamer to impair Hsp70-mediated clearance of misfolded proteins and simultaneously target ERα for ubiquitination/degradation. This tactic of one molecule-mediated dual targeting to simultaneously inhibit the general cancer-promoting Hsp70 system and eliminate a cancer-type-specific protein factor such as ERα could be further developed into an effective strategy for cancer therapy.
In addition, our study sheds light on the mechanism of Hsp70-mediated protein triage. Inhibition of CHIP-mediated ubiquitination by the monofunctional AptHsp70-1 and the reversal of the effect by the bifunctional aptamer indicate a direct correlation between the extent of CHIP-mediated substrate ubiquitination and the strength of Hsp70-substrate interaction. Paradoxically, interaction of CHIP with Hsp70 inhibits Hsp70 ATPase activity and destabilizes Hsp70-substrate interaction [10]. Based on our study, we propose a model in which destabilization of Hsp70-substrate interaction by CHIP may function as a built-in negative feedback on CHIP-mediated ubiquitination of Hsp70 substrates (Fig. 5B). In this feedback system, CHIP is both the signaling molecule (executor of ubiquitination) and also the direct negative regulator of an upstream pathway component (Hsp70 activity). Therefore, both the signaling and the feedback occur on the same timescale, resulting in a strong negative feedback system that cannot produce a transient response [47]. So, a stimulus such as stress or heat shock that causes proteins to misfold and activates the Hsp70 chaperone system would not result in an instant and transient surge in CHIP-mediated ubiquitination of Hsp70 substrates. Instead, it would result in a slow increase in ubiquitination, which would persist as long as the stimulus is present [47].
Such a mechanism would prevent an outburst of chaperone substrate degradation, which is costly in energy, but at the same time allows for sustained degradation of extensively misfolded proteins, which are likely to have a stronger interaction with Hsp70. The existence of this control mechanism may enable the Hsp70 chaperone system to efficiently triage extensively misfolded proteins for degradation without jeopardizing the recovery of misfolded, but repairable, substrates. Disruption of this delicate balance could result in diseases. For example, cystic fibrosis is caused by loss of cystic fibrosis transmembrane conductance regulator protein (CFTR), an Hsp70 substrate. The cystic fibrosis-associated mutant, CFTRΔF508, is folding competent, but its folding kinetics is slower than wild type, which causes increased Hsp70-CHIP-mediated CFTRΔF508 degradation [48–50]. Therefore, manipulation of the Hsp70 chaperone system may alleviate such loss-of-function protein misfolding disorders.
Footnotes
Acknowledgments
This work was supported by a grant (R01CA140730) from the National Institutes of Health and a Research Scholar Grant (RSG-09-159-01-CDD) from the American Cancer Society to H.S.
Author Disclosure Statement
No competing financial interests exist.