
Abstract
The very common GNB3 c.825C>T polymorphism (rs5443) is present in approximately half of all human chromosomes. Significantly, the presence of the GNB3 825T allele has been strongly associated with predisposition to essential hypertension. Paradoxically the presence of the GNB3 825T allele, in exon 10, introduces a pathogenic alternative RNA splice site into the middle of exon 9. To attempt to correct this pathogenic aberrant splicing, we, therefore, bioinformatically designed, using a Gene Tools® algorithm, a GNB3-specific, antisense morpholino. It was hoped that this morpholino would behave in vitro as either a potential splice blocker and/or exon skipper, to both bind and inhibit/reduce the aberrant splicing of the GNB3 825T allele. On transfecting a human lymphoblast cell line homozygous for the 825T allele, with this antisense morpholino, we encouragingly observed both a significant reduction (from ∼58% to ∼5%) in the production of the aberrant smaller GNB3 transcript, and a subsequent increase in the normal GNB3 transcript (from ∼42% to ∼95%). Our results demonstrate the potential use of a GNB3-specific antisense morpholino, as a pharmacogenetic therapy for essential hypertension.
Introduction
T
These different heterotrimeric G proteins, in turn specifically bind to different G protein-coupled receptors (GPCRs), which are stimulated by specific ligands or light. Following activation of these GPCRs, this subsequent change in confirmation activates the bound heterotrimeric G protein [3]. As only one of the five β subunits selectively binds with the many different α (22) and γ (14) subunits, this results in a plethora of structurally different heterotrimeric G proteins. Many of these different heterotrimeric G proteins will also have different pathway specificities in the many different cell types of vertebrates [4], for example, GNB3 is the only one of the five beta subunits that is expressed in adult cone cells, but is not expressed in rod cells where GNB1 is expressed [5]. Moreover, in early development, GNB3 expression is largely restricted to the eye [6]. Later in development, however, the GNB3 gene is more ubiquitously expressed throughout the vertebrate body and is often coexpressed, although at a lower level [6], with GNB1 and possibly the other GNB genes in many of the bodies vital organs, for example, brain, heart, liver, and kidney [7–11].
Gβ3, together with particular α and γ subunits, therefore, selectively binds to a significant number of the thousands of different GPCRs, which are encoded by the 800 genes in the human genome [12].
Given the wide range of tissues that the GNB3 gene is expressed, it is therefore not surprising that both the GNB3 knockout mouse model [13] and the GNB3 p.D153del homozygous, retinopathy globe and glomerulus enlarged (rgage/rge) chicken, have been shown to suffer from both retinal defects, and other pleiotropic phenotypes [7,14]. For example, in both of these homozygous GNB3 mutant organisms, there has been reported a high embryonic/infant mortality, for example, only ∼10% of the homozygous GNB3 knockout mice survived for longer than 3 weeks after birth [13]. Similarly, we previously reported a ∼30% embryonic lethality in the homozygous p.D153del chicken [7]. In the latter organism initially only a severe retinopathy globe enlarged (rge) phenotype was detected [15]. Later studies, however, by our research group discovered that the p.D153del eight-month-old chickens, also suffered from enlargement of the glomerular capsule, causing glomerulomegaly and tubule-interstitial inflammation, which causes botha retinopathy globe and glomerular-enlarged (rgage) phenotype [14]. These results suggest that the signaling pathways involving GNB3 are vital for both the normal development and maintenance of both the eye and the kidney's renal cortex. In contrast, other GNB3-expressing selected tissues, taken from homozygote c.D153del GNB3 chickens, that is, brain, heart, liver, and pancreas, appeared to be histologically unaffected, despite the finding of significant alterations in the signaling pathways in these tissues [14].
Recently, however, it has also been shown that GNB3 knockout mice suffering from cone function defects have also been shown to suffer from bradycardia [6]. These mice surprisingly, however, showed no other differences in metabolic phenotypes, for example, blood pressure and body weight compared to normal age-matched controls. Moreover, the decrease in heart rate appeared to be due to changes in neural signaling, as isolated GNB3 KO mouse hearts showed no difference to isolated normal hearts, following standard cardiac ion channel functional tests [6].
No significant phenotypic alterations have, to date, been detected in either the heterozygous GNB3 knockout mice or the heterozygous c.D153del chicken, suggesting that relatively normal signaling pathways in most tissues are possibly maintained with only 50% of GNB3 levels. Related to these findings, it has recently been found that both humans and transgenic mice that possess an extra copy of the GNB3 gene have been shown to exhibit an obese phenotype [16]. Taken together, these results suggest that altering GNB3 expression by either haploinsufficiency or by duplication, but probably not by complete ablation, is likely to change both normal metabolism and possibly dietary behavior in humans.
To date, no humans have so far been reported with an equivalent GNB3 inactivating mutation, similar to either our chicken D153del mutation or with a deletion knockout mouse-like mutation. However, the very common GNB3 gene variant c.825C>T (rs5443), which is present in approximately half of all human chromosomes, has been shown to be significantly associated with an increased risk of hypertension, especially in obese patients [17–19], regardless of the patient's ethnicity. Interestingly, the global allele frequency distribution of the GNB3 825T allele is remarkably similar to that of common polymorphisms in salt-controlling genes, for example, the Angiotensin I-converting enzyme (ACE), angiotensinogen (AGT), and both the sodium channel, nonvoltage-gated 1 alpha and gamma subunits (SCNN1A/ENACα and SCNN1G/ENACγ) [20–23]. The GNB3 825T allele and particular functional alleles (eg, the ACE D allele) in these other salt-regulating genes have significantly higher frequencies in Sub-Saharan tropical Africa than in any other parts of the world, where the GNB3 825T allele appears to be strongly selected against. In contrast, these data also strongly suggest that the common Sub-Saharan alleles were strongly selected before mankind's ancestors migrated out of Africa, ∼100,000 years ago [22,21]. The most likely reason for the differential selection of the 825T GNB3 allele and other salt-controlling alleles, in these different climes, is due to the hot and wet climate and low salt availability, endured by humans in central Africa at this time. In addition, the sub-Saharan hot climate inevitably led to the selection of humans with an increased capacity to sweat and cool down through evaporation. This selection has in turn also led to an increase in the frequency of gene alleles, for example, GNB3 825T that increases renal sodium conservation, to combat this inevitable loss of both salt and water. GNB3 and the 825T allele in particular is, therefore, likely to be involved in GPCR-stimulated pathways that involve the control of urinary salt uptake, for example, through the angiotensin/NHE [19], bradykinin, and/or SCNN/ENAC pathways [24]. However, recent experiments using the GNB3 knockout mice, which were infused with angiotensin II, with no physiological changes in blood pressure, suggest that GNB3 is probably a peripheral player in this particular pathway [6]. Interactions, however, between variant haplotypes in the obesity-related FTO/IRX3 locus [25] and the 825T allele of GNB3, have recently been shown to help contribute to the varied clinical phenotypes in hypertension [26].
Significantly, the GNB3 825T allele has also previously been implicated in obesity [23], cardiovascular disease [27–33], low birth weight [34], Alzheimer's disease, cognitive decline [35], erectile dysfunction [36–40], tumor progression [41–46], and as a genetic marker for drug response [47–56].
The mechanism by which the GNB3 825T allele causes carriers to be predisposed to hypertension and other diseases, however, still remains to be fully elucidated. The GNB3 825T allele, unlike the 825C allele, paradoxically introduces an alternative RNA splice site into the middle of exon 9, possibly by altering the GC ratio critical to both nucleosome methylation and spliceosome binding, during gene expression [18]. The GNB3 gene with an 825T allele, therefore, produces two different transcripts in an ∼50:50 ratio (Fig. 1). The normal splicing event 8–9a, produces a transcript that is 123 bp longer than the aberrant pathogenic 8–9b transcript. The former 8–9a transcript codes for the normal stable Gβ subunit, whereas the latter 8–9b translates into a highly unstable and possibly pathogenic isoform Gβ3s, which is deleted for 41 highly conserved amino acids [57]. In addition, as a result of this alternative splicing event, the expression of the normal 8–9a transcript is reduced by ∼50%, compared to GNB3 genes carrying the 825C allele (Fig. 1).

This diagram shows the proposed mechanism of action of the GNB3 antisense morpholino (M) and its binding site, which spans both sides (9a and 9b) of the cryptic splice site, in the middle of exon 9.
Finding a specific drug to specifically try to correct the hypertensive and possibly some of the other GNB3 825T allele-associated phenotypes is, therefore, an attractive proposition. One approach chosen in an attempt to specifically correct the analogous missplicing of the pathogenic common hemoglobin (HbE) allele, HBB c.79G>A (p.E27K), was to use an antisense morpholino, to specifically target its pathogenic missplicing [58]. This antisense morpholino treatment of HeLa cells, expressing the HbE allele, resulted in a ∼70% increase in the normal splice product. More recently antisense exon skipping morpholinos have also been successfully used to treat Duchenne muscular dystrophy patients, who suffer from out-of-frame forming exon deletions. Such patients demonstrated a dramatic improvement in their clinical symptoms, with apparently little side effects following intravenous treatment, using apparently very stable and long-lasting antisense morpholinos [59].
To attempt to combat either or both of these potentially pathogenic events in hypertensive patients, carrying an 825T allele, we therefore used the Gene Tools® algorithm to design a so called exon skipping morpholino, to try to target and correct the aberrant splice site, in the middle of exon 9, in the GNB3 gene (Fig. 1). In addition, we also designed an ordinary antisense DNA oligonucleotide to try to block the aberrant GNB3s transcript. Both of these oligonucleotides were then used to transfect two different human lymphoblastoid cell lines, with two different GNB3 genotypes, namely homozygous 825T and heterozygous 825T/825C.
Materials and Methods
Morpholino and antisense oligo design
The following aberrant splice site GNB3 gene sequence, in the middle of the Exon 9 gene, was submitted to the Gene Tools algorithm (www.gene-tools.com) for exon skipping morpholino, design:
ctcttcatttcgggggcctgtgatgccagTGCCAAGCTCTGGGATG TGCG.
This algorithm then designed the following 25 bp morpholino (M) sequence, which is complementary to the GNB3 aberrant splice site:
5′-CACTGGCATCACAGGCCCCCGAAAT-3′, which was then ordered from Gene Tools. In addition, a allele specific (AS) oligonucleotide 5′-GAGCTTGGCACACGTGGTGT-3′, which is complementary to the last 10 bases in exon 8 and the first 10 bases after the aberrant “AG” splice site in exon 9, was also ordered from Sigma-Aldrich. This oligonucleotide was designed to hopefully bind preferentially to the GNB3s mRNA transcript and less to the normal GNB3 transcript.
Polymerase chain reaction primers
The following reverse transcriptase (RT-PCR) primers were designed using Primer 3 software and ordered from Sigma: GNB3 exon 7–10 forward GAGCTTTCTGCTCACACAGG, GNB3 exon 7–10 reverse TCATGGAGTCCCAGACATTG-3′, GNB3 full coding with HindIII linker forward GCAAGCTTGCCATGGGGGAGATGGAGC, GNB3 full coding with Xho1 linker reverse CTCGAGTCAGTTCCAGATTTTGAGGAA GCTG.
Actin forward primer GCAAAGACCTGTACGCCAAC, and actin reverse primer 5′-CGTCATACTCCTGCTTG CTG-3′.
Cell culture
The GM18500 GNB3 825T/825T (TT), GM18506 GNB3 825T/825C (CT), and 825C/825C (CC) (GM19116) Nigerian Yorba tribe, transformed, lymphoblastoid cells were obtained from Coriell Cell Repositories (http://ccr.coriell.org/).
The cells were added drop by drop into a T-50 culture flask (50-mL) containing 20 mL of Roswell Park Memorial Institute–1640 (RPMI 1640) culture medium (Invitrogen), which was then agitated to ensure even spreading. Each of these flasks was then incubated at 37°C, for 24 h. The cells were then removed after incubation and their confluence checked under the microscope. Once confluent, the cells were subcultured by removing the old medium and washing with phosphate-buffered saline (PBS), to remove the serum.
After incubation, 20 mL of cells in RPMI-1640 media were added to T-75 culture flasks, and incubated overnight at 37°C, 5% CO2. A 30 mL of preheated RPMI-1640 media was added to top the cells up to 50 mL, leaving them to incubate for around 48–72 h until the pH of the media had decreased. The cultures were manually agitated daily to break up the cell clumps formed.
Transfection (lipofectamine™)
When the cells had reached 50%–70% confluence, they were shaken to break up clumps, transferred into 15-mL centrifuge tubes and centrifuged at 720 g for 8min, removing the supernatant. The pellet was washed twice, by resuspending the pellet in 10 mL PBS, centrifuging the cell suspension at 720 g for 8 min, and removing the supernatant. The cells were then resuspended in 2 mL of Opti-MEM®, and 30 μL of the suspension was removed so the cells could be counted using a hemocytometer. Once the cells were counted, a 500 μL dilution of each suspension was plated in a six-well multiple well plate at a density of ∼3.6 × 106 cells per well, containing 2 mL of Opti-MEM. The six-well plate was then incubated for 2 h at 37°C, 5% CO2, to starve the cells of serum before transfection. Four microliters of the GNB3 morpholino (1 mM/μL) and 4 μL antisense oligonucleotide (1 mM/μL) were both initially mixed with 200 μL Opti-MEM and left at room temperature for 10 min. These DNA/Opti-MEM mixtures were then added to another mixture containing 12 μL Lipofectamine and 200 μL Opti-MEM, which had also been preincubated for 10 min.
To measure the transfection efficiency, 3 μL of green fluorescent protein (GFP)-expressing plasmid was also added to the mixture. The two mixed tubes were then incubated for a further 20 min, allowing the Lipofectamine to form a complex with the morpholino and antisense oligonucleotide. One hundred microliters from the morpholino/Lipofectamine tube was added to the four morpholino wells, and 100 μL from the antisense/Lipofectamine tube was added to the four antisense wells. For the four mock samples 12 μL Lipofectamine was mixed with 200 μL Opti-MEM, incubated for 10 min, then 3 μL of GFP was added and allowed to incubate at room temperature for 20 min before being added to the mock cells. The six-well plate was gently shaken back and forth to ensure distribution of the Lipofectamine complex. Another 1,000 μL of Advanced RPMI 1640 was added to each of the 12 wells. These were placed in the incubator at 37°C overnight (∼24 h). Afterward, the six-well plates were removed from incubation and 3 mL of RPMI 1640 was then added to each well. The six-well plates were then incubated for a further 48 h at 37°C. The six-well plate was shaken gently back and forth to ensure distribution of the Lipofectamine complex. Another 1,000 μL of Advanced RPMI 1640 was added to each of the 12 wells. These were placed in the incubator at 37°C overnight (around 24 h). Afterward, the six-well plates were removed from incubation and to each of the 12 wells, 3 mL of RPMI 1640 was added. The six-well plates were then incubated for a further 48 h at 37°C.
RNA extraction
The medium containing cells from each well was centrifuged and resuspended in 1 mL of PBS and transferred to a 1.5-mL tube and centrifuged at 425 g for 10 min, before the supernatant was removed. The 1.5-mL tubes were placed on ice and to each 100 μL QIAzol® (Qiagen) was added and mixed by pipetting until the cell pellet was fully resuspended; the tubes were then incubated at room temperature for 5 min, before being placed back on ice. To each 1.5-mL tube, 20 μL of chloroform was added and vortexed for 10 s, then incubated for 3 min at room temperature, before being placed back on ice. The 1.5-mL tubes were then centrifuged at ∼23,000 g for 15 min at 3°C. After this, each 1.5-mL tube presented three phases: a top colorless aqueous phase, a middle white interphase, and a lower red phenol–chloroform phase. The upper aqueous phase containing the RNA was removed from each 1.5-mL tube without disturbing the two lower phases and added to a new labeled 1.5-mL tube. The RNA was precipitated by adding 50 μL of isopropanol and incubated for 10 min at room temperature. The1.5-mL tubes were centrifuged at ∼23,000 g for 10 min at 3°C. The supernatant was removed and the pellets were washed by mixing with 100 μL of 75% ethanol, and centrifuged at 239 g for 10 min at 3°C, the supernatant was removed and this wash step was repeated. Afterward, the RNA pellet was left for the ethanol to evaporate for 7 min. The RNA samples were then further purified to ensure no genomic DNA contamination using an RNeasy Mini Prep Kit, Qiagen, using the manufacturer's protocol. RNA was then quantified by spectrophotometry and diluted to a concentration of 100 ng/μL.
RT-PCR
cDNA synthesis was undertaken using the manufacturer's instructions using the Oligo dT method in an M-MLV Reverse Transcriptase Kit (Life Technologies). Polymerase chain reaction (PCR) was carried out using a MyTaq™ Red Mix (Bioline) PCR Kit in a 50 μL final volume, containing 100 ng cDNA and 0.2 μM of forward and reverse primers. Thermocycling was carried out with an initial denaturation at 95°C for 5 min, followed by 35 cycles of 94°C for 30 s, 45°C for 45 s, and 72°C for 45 s. A final extension was carried out at 72°C for 45 s. PCR samples were loaded on a 2% gel containing 1X TAE buffer and 10 μL Save View™, ABM. Bioline HyperLadder II was used as a size marker and the gel was run at 150 V for 90 min. Semiquantitative analysis of the JPEG image of the gel was carried out using ImageJ software (http://imagej.nih.gov/ij/).
Protein extraction
Coriell cell lines, possessing the following GNB3 genotypes 825CC (GM19116), 825CT (GM18506), and 825TT (GM18500) cells, were either mock or antisense morpholino transfected. After 24–48 h incubation, the cell lines were checked for 70%–90% confluence in their wells. Once this was achieved, the cells were transferred into a 15-mL centrifuge tube and pelleted at 720 g. The supernatant was discarded and the pellet was resuspended in 5 mL PBS to wash, the cells were then centrifuged at 720 g to pellet, discarding supernatant, repeating once. The cells were resuspended in 150 μL of ice-cold RIPA buffer, mixed by pipetting, then placed directly on ice, and carefully transferred into a labeled 1.5-mL microcentrifuge tube, then incubated for 5 min. The microcentrifuge tubes were centrifuged at 28,000 g for 10 min at 4°C. Ten microliters of the supernatants containing the protein lysate was transferred to fresh microcentrifuge tubes for analysis of concentration, by the Bradford Assay. The remaining volumes were measured (∼140 μL) and added to fresh microcentrifuge tubes, adding 1:1 sample buffer (containing β-mercaptoethanol) and mixed by pipetting. The samples were heated at 94°C for 15min shaking at 900 rpm, and the samples were centrifuged at 28,000 g for 10 min. The supernatants were transferred in to fresh microcentrifuge tubes and aliquots stored in a −80°C freezer.
Western blot (WesternBreeze® Kit)
The Ab154866 Gβ3-specific antibody was obtained from Abcam. It is a rabbit monoclonal antibody raised against a 14 amino acid residue synthetic peptide taken from within the region 100–150 of human Gβ3 protein. The antibody was diluted in a diluent blocking solution provided in the WesternBreeze Kit (7 mL Filtered dH20 + 2 mL Blocker A +1 mL Blocker B), Ab154866 was diluted 1:1,000 by adding 10 μL of antibody to 10 mL of diluent.
The thawed protein lysate samples were reheated at 94°C, 900 rpm for 10 min, and then centrifuged for 1 min 5,000 g. The sodium dodecyl sulfate–polyacrylamide gels were placed into the XCell SureLock® Mini-Cells filled with 1× Tris-glycine running buffer and the wells loaded with 10 μg of protein, according to the Bradford Assay results, and 5 μL of SeeBlue® Prestained Standard protein marker from Life Technologies was also added. The lid was attached and set to 200 V and 200 mA, allowing the gel to run for 1 h 30min. Blots were then probed with either of the two antibodies, according to the manufacturer's instructions.
Results and Discussion
As can be seen from both Figs. 2 and 3, there appears to be significant inhibition of the formation of the aberrant 8–9b splicing, in both the homozygous 825TT cell line (reduced from ∼58% to ∼5%) and the heterozygous 825CT cell line, following transfection with the GNB3-specific antisense morpholino. Moreover, following morpholino transfection of the 825TT cell line, there also appeared to be a significant increase in the production of the normal 8–9a transcript (increased from ∼42% to 95%). In contrast from Fig. 2, it can be seen that both mock transfection and transfection with the allele specific unmodified (AS) oligonucleotide of the 825TT cell line, appears to have little effect in altering the expression of neither the 8–9b nor the 8–9a splicing event. Encouragingly, mock transfection or transfection with either the morpholino or the allele specific (AS) morpholino affected the expression of the control actin gene.

This diagram shows the semiquantitative (reverse transcriptase) RT-PCR results for both the 825TT and 825CT cell lines. Lane 1 contains the Bioline 100 bp (HyperLadder) marker. Lanes 2–7 are reverse transcriptase (RT-PCR) results generated using the full-length GNB3 primers as described in the methods. The expected size of the full-length product is 1,026 bp for the 8–9a normal transcript and 903 bp for the alternative 8–9b pathogenic transcript. Lanes 8–13 are RT-PCR bands generated with exon 7–10 primers and generate an expected band of 490 bp for the 8–9a normal transcript and 367 bp for the pathogenic 8–9 b transcript. Lanes 14–19 are RT-PCR products generated from the actin-specific primers. Lanes 2, 5, 8, 11, 14, and 17 are RT-PCR products generated from mock-transfected GNB3 825 TT or TC cell lines. Lanes 3, 6, 9, 12, 15, and 18 are RT-PCR products generated from GNB3 antisense morpholino (M)-transfected GNB3 825 TT or TC cell lines. Lanes 4, 7, 10, 13, 16, and 19 are RT-PCR products generated from GNB3 allele specific (AS) unmodified oligonucleotide transfected GNB3 825 TT or TC cell lines.
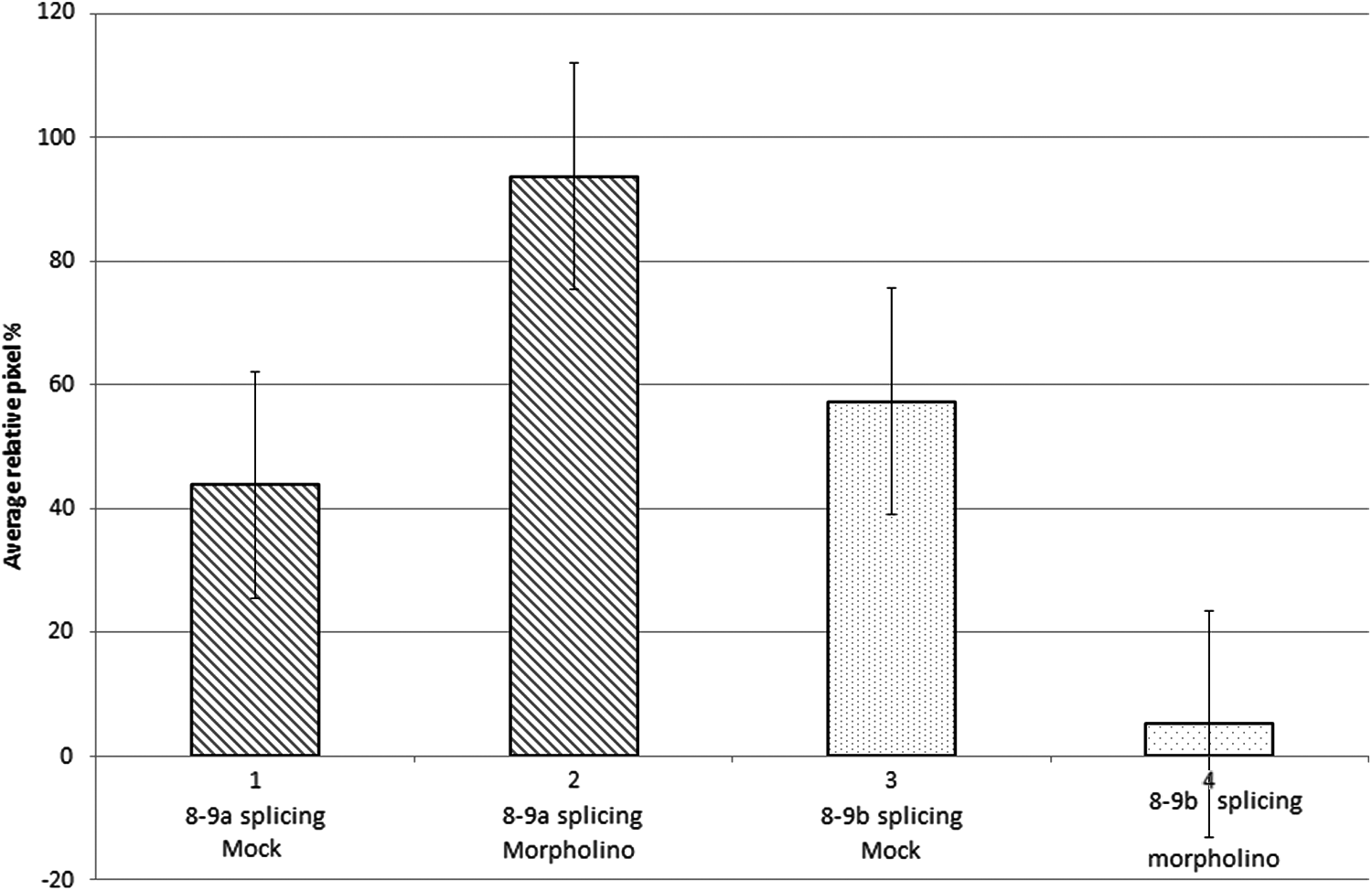
This diagram represents the ImageJ relative average% pixel analysis, with standard error bars, of lanes 2, 3, 8, and 9 shown in the RT-PCR semiquantitative gel, in Fig. 2. Block 1 represents the average% pixels from the 8–9a splicing event with mock treatment, lanes 2 and 8 in Fig. 2. Block 2 represents the average% pixels from the 8–9a splicing event with GNB3 antisense morpholino (M) treatment, lanes 3 and 9 in Fig. 2. Block 3 represents the average% pixels from the pathogenic 8–9b splicing event with mock treatment, lanes 2 and 8 in Fig. 2. Block 4 represents the average% pixels from the pathogenic 8–9b splicing event with antisense morpholino (M) treatment, lanes 3 and 9 in Fig. 2.
These initial RNA results suggest that our GNB3 antisense morpholino appears to significantly correct both of the potential pathogenic effects of the GNB3 825T allele, first downregulation of the aberrant 8–9b, and second, upregulation of the normal 8–9a splicing event.
From the Gβ3 western blot results shown in Figs. 4 and 5, however, it can clearly be seen that only the normal Gβ3 splice variant and not the unstable and short-lived Gβ3s protein is detected with the Gβ3 monoclonal antibody Ab154866. This is despite this antibody being raised from a portion of the Gβ3 protein, amino acids 100–150 that is present in the truncated Gβ3s protein, which is deleted for amino acids 168–207. This result is similar to the one achieved by Sun et al. [57], who were also unable to detect the unstable Gβ3s protein in their experiments. It is, therefore, impossible to determine from this western blot, whether our antisense morpholino is actually downregulating the amount of the Gβ3s protein, as indicated in our RNA results. Moreover, contrary to our RT-PCR results in Fig. 2, there appears to be no significant difference in the quantity of the normal Gβ3 protein, in both the 825TT and 825CC cell lines, following treatment with the antisense morpholino.
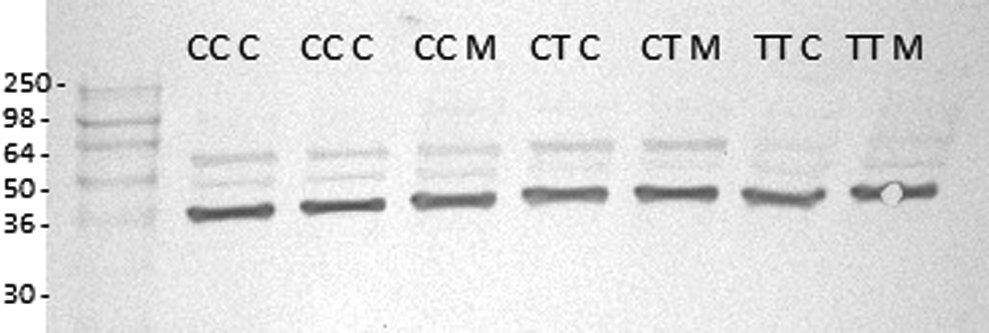
This western blot shows, homozygous 825 CC, heterozygous CT, and homozygous TT cell lines, which were either transfected morpholino (M) or mock-transfected control (C). The extracted protein samples from these treated cell lines were blotted and probed using the specific anti-Gβ3 antibody Ab154866.
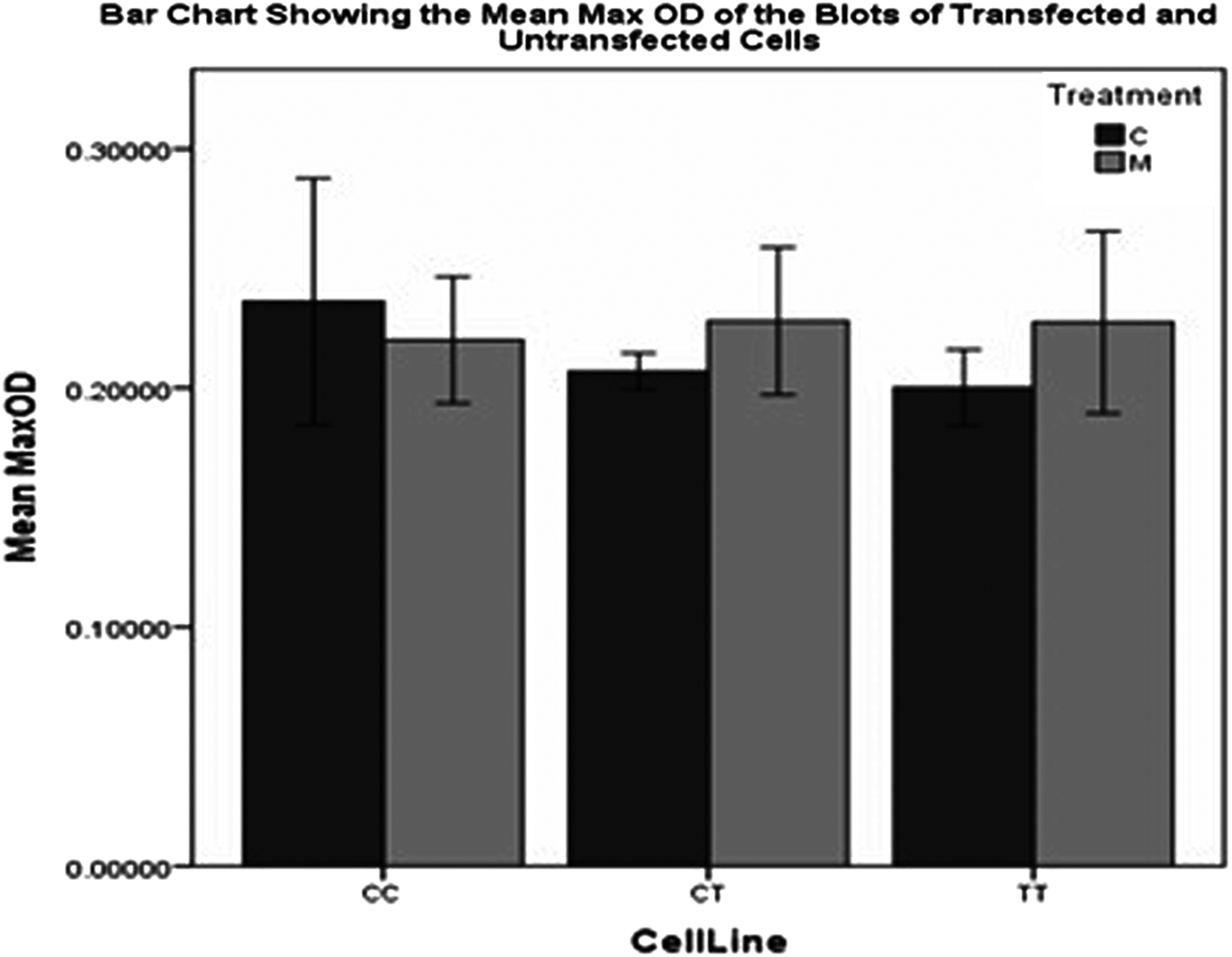
This bar chart shows the mean MaxOD of the Gβ3-specific bands in the western blot shown in Fig 4.
The semiquantitative RT-PCR results obtained in our experiments, however, still suggest that the GNB3 antisense morpholino may still be useful in helping lower blood pressure in both normal and obese individuals who carry the 825T allele [17]. This is because some or all of the pathogenic effects of the 825T allele are likely to be due to the presence of the unstable Gβ3s protein subunit, rather than the mere reduction of the expression of the normal Gβ3 subunit. In support of this argument, it has recently been found that many different specific deleterious point mutations in the related GNB1 and GNB2 genes are able to transform cell lines from being cytokine dependant, to a cytokine-independent phenotype [60]. Moreover, these same mutations have been found in many different human cancers. These findings strongly suggest that many different mutations in the heterotrimeric C protein β subunits, including the GNB3 splice variant Gβ3s, are likely to cause dominant/negative effects on both βγ and α subunit G protein signaling pathways [61]. If this dominant/negative effect does ultimately prove to be the main predisposing hypertension disease mechanism for the GNB3 825T allele, then antisense morpholinos, inhibiting the formation of the Gβ3s subunit, may have great therapeutic value, especially in obese hypertensive patients [17].
Footnotes
Acknowledgments
The authors are grateful to both Janet Horrocks and Scott Cameron of Abertay University for their expert technical assistance in western blotting.
Author Disclosure Statement
J.M. is an employee of NHS Scotland, T.H. is an employee of Queen Mary University of London, and D.L. is an employee of the University of Abertay. None of these authors has any known conflicts of interest.