
Abstract
miRNAs are highly conserved class of small ncRNAs whose involvement in human pathophysiologies is extensively investigated. MiR-21 is a well established oncogenic miRNA whose deregulation plays a significant role in onset and progression of cancer. The need of novel approaches to downregulate miR-21 is rapidly expanding. Potent inhibition of miR-21 is achieved by chemically modified 2′-O-methyl RNA oligonucleotide. The serinol capping at 3′ and 5′ends and the interspersed 2′-O-(R-2-amino-3-methoxypropyl) uridine units enhance the nuclease resistance and efficacy of 2′-O-methyl RNA for the inhibition of miR-21. This represents a simple and novel modification for developing oligonucleotide-based therapeutics.
Introduction
M
Antisense technologies that use synthetic reverse complements have been extensively used to suppress or knockdown the expression of target RNAs. Antisense inhibition of miRNA has been an important tool to uncover miRNA functions. Precise functions of individual miRNAs have been delineated by loss of function studies using various anti-miRNA oligonucleotides (AMOs), which either degrade or sequester the miRNAs making them unavailable to bind to their target mRNAs leading to depression of their function. Furthermore, AMOs have been used as probes for detection [5–7], target validation, functional characterization, and therapeutic intervention of miRNAs [8,9]. AMOs are designed to bind to complementary miRNA through Watson–Crick hybridization and thereby repress the activity. Despite this seemingly straightforward and simple approach of sequence-specific targeting of miRNA, unmodified AMOs with phosphodiester backbone present several shortcomings such as poor cellular uptake, rapid degradation by nucleases in serum, low-target hybridization efficiency, and nonspecific targeting. The challenge still remains to bestow AMOs with adequate pharmacodynamic and pharmokinetic “drug-like” properties for therapeutic strategies. This can be successfully brought about by incorporating various chemical modifications in the nucleobase, internucleotide linkages, or in sugars to increase target binding affinity, specificity, potency, and biostability of AMOs [10]. These modifications include phosphorothioates (PS), 2′-O-methoxyethyl (MOE) [11], 2′-O-methyl (2′-OMe) [12,13], 2′-fluoro [14], peptide nucleic acids (PNAs) [15,16], and locked nucleic acids (LNAs) [17,18]. Although these modifications confer high duplex stability while binding with the target RNA, there is a need to further improve their enzymatic stability such that they do not get degraded by endogenous nucleases. Earlier reports suggested that 2′-O-methyl RNA oligonucleotides can provide efficient and simple way to block small RNA function in vivo [12,19]. Recently, terminally modified 14 mer 2′-O-methyl RNA has been shown to inhibit miRNA function with improved mismatch discrimination [20]. The modifications include trimethoxystilbene residue and three 2′-fluoro-2′-deoxynucleotides at the 5′ and 3′ terminal, respectively [20]. 2′-O-methyl RNA is a naturally occurring modification that confers high binding affinity (enhanced melting temperature, Tm) with the target RNA making stable duplexes [21]. In addition to having high affinity, an ideal AMO should also be resistant to both endo- and exonucleases present in the serum. 2′-O-methyl RNA, owing to its chemistry, impedes the ability of endonucleases to degrade single stranded oligonucleotides, but has the limitation of being sensitive to serum exonucleases [22]. A recent report used a non-nucleotide modifer, N,N-diethyl-4-(4-nitronaphthalen-1-ylazo)-phenylamine (“ZEN”), which improves the efficacy when placed at or near the ends of 2′-O-methyl RNA [23]. In our attempt to enhance nuclease resistance with conserved binding affinity of 2′-O-methyl RNA oligonucleotides, recently we described a novel chemical modification comprising methoxy- and amino-groups, that is, 2′-O-(R-2-amino-3-methoxypropyl) (R-AMP) substitutions [24]. Furthermore, we reported the enhanced nuclease resistance of these oligonucleotides by capping the 3′ and 5′ ends with serinol units. The presence of interspersed UR-AMP units in 2′-O-methyl RNA oligonucleotide capped with serinyl units (S) at 3′/5′ ends was found to be superior to the unmodified 2′-O-methyl RNA, 2′-O-methyl RNA capped at 3′/5′ ends by serinyl units, as well as LNA-OMe mixmers, when used in a cell-based splice correction assay [25]. These promising results prompted us to investigate the efficacy of these modifications (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat) in 2′-O-methyl RNA oligomers as inhibitors of miRNA in cell culture–based system. In this study, we report our modified oligonucleotides as potent miRNA inhibitors. The chemical synthetic routes to the modified uridine phosphoramidite monomer for introducing (UR-AMP) [24] and serinyl-end cap [25] were simple and affordable (Supplementary Figs. S2 and S3).
These monomers were used in the automated DNA synthesizer along with 2′-O-methyl monomers to synthesize modified 2′-O-methyl RNA oligonucleotides (Fig. 1 and Supplementary Table S1).
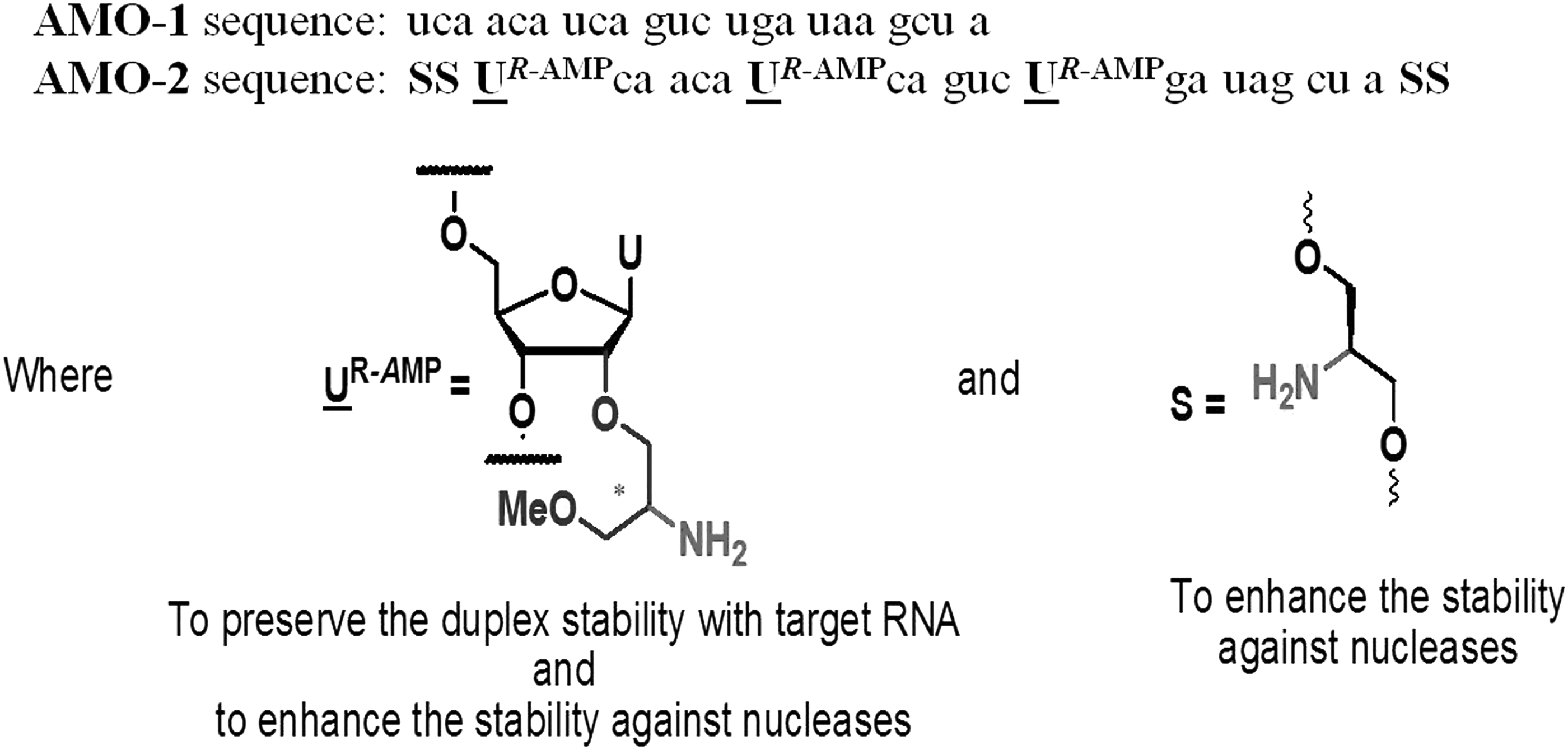
The sequence and modifications of AMOs against miR-21. AMO-1 and AMO-2 are uniformly 2′-O-methyl modified RNA reverse complementary to miR-21. AMO-2 harbors three UR-AMP monomers positioned at 2, 8, and 14 and two non-nucleotidic Serinyl caps at both termini. AMOs, anti-miRNA oligonucleotides.
Materials and Methods
Design and synthesis of anti-miR-21 oligonucleotides
The oligonucleotides were synthesized on a BioAutomation MM4 DNA synthesizer using amidites
The primary objective for the design of the AMOs was to impart nuclease resistance without compromising target affinity; we chose to include PS modification (well characterized for imparting nuclease resistance) as a control for comparing the potency of the AMOs. In PS modification, nonbridging oxygen in the phosphate backbone is substituted by a sulfur atom. This hinders the ability of nucleases to degrade these bonds. PS modifications have also been used successfully in the FDA-approved oligonucleotide drug, Formivirsen [26]. Thus, 2′-OMe RNA with full phosphorothioate backbone was used as another control. For the negative control, 2′-OMe RNA scrambled sequence was used: 5′ CAGUACUUUUGUGUAGUACAA 3′. All the comparisons were made to the chemistry matched scramble control. These two sequences were purchased from Sigma-Aldrich Co.
Cell culture and transfection
MCF-7 (human breast cancer cell line) and MDA-MB-231 (human breast adenocarcinoma) cells were cultivated in Dulbecco's modified Eagle's minimum essential medium (DMEM) high-glucose GlutaMAX™ (Life Technologies) supplemented with 10% heat inactivated fetal bovine serum and fetal bovine serum (Life Technologies) and were maintained at 37°C in 5% CO2 humidified incubator. One day before transfection, cells were seeded in 6-well plates, 24-well plates, or 96-well plates to attain ∼60%–70% confluency. All transfections were performed using Lipofectamine 2000 (Invitrogen) using the manufacturer's guidelines.
Dual luciferase assay
To compare the functional potency of all AMOs against miR-21, a dual luciferase screen was performed. By cloning the inverted complement of miR-21 sequence under the renilla gene, we created a screening tool to assess the activity of miRNA and its inhibitors. The reporter plasmid (psiCHECK™-2 Vector; Promega) expresses renilla gene and a firefly gene for normalization of signal accounting for variability in transfection. The miR-21 target encoding two repeats of reverse complement of miR-21 is cloned between XhoI and NotI cloning site downstream of the renilla translation stop codon. The sequence repeats used to clone target downstream of renilla gene is: 5′ TCAACATCAGTCTGATAAGCTA
Quantitative real-time reverse transcriptase–polymerase chain reaction
The MCF-7 cells were seeded in six-well plate (3 × 105 cells/well), grown to 60%–70% confluency, and transfected with 50 nM each of PS/AMO-1/AMO-2. Cells were then incubated at 37°C for 24 h in high-glucose DMEM in 5% CO2. The media was removed, and cells were washed with 1× phosphate-buffered saline (PBS), and total RNA was isolated using TRIzol® Reagent (Invitrogen). Expression levels of mature miR-21 were determined by QuantiMir Kit (Catalog No. RA660A-1; SBI). Forward primer used for quantitative real-time reverse transcriptase–polymerase chain reaction (qRT-PCR) was 5′ TAGCTTATCAGACTGATGTTGA 3′. The universal reverse primer was provided by the SBI Kit. The microRNA expression levels were detected using SYBR-Green I PCR Master Mix (Applied Biosystems) on Roche LightCycler LC 480 and normalized with respect to U1 as a reference gene. The real-time data analysis was performed by ΔΔCt method [27].
Western blot
Total protein was extracted from transfected and untransfected MCF-7 cells using CelLytic M buffer (Sigma-Aldrich Co.). The cellular debris was discarded by refrigerated centrifugation, and the supernatant containing the cellular lysate was transferred to a fresh tube. Protein concentration for each sample was estimated using BCA™ protein assay (Pierce Chemical Co.). An equivalent amount of protein from each sample was loaded on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Following transfer to nitrocellulose membrane, the blot was blocked with 5% bovine serum albumin for 2 h at room temperature. Next, the blot was incubated with mouse primary antibody (1:1,000) specific for programmed cell death 4 (PDCD4) and phosphatase and tensin homolog (PTEN) target proteins (Abcam). The other half of the membrane was incubated with primary antibody (1:2,000) specific to GAPDH, which was used as a loading control. After washing the blot thrice with 1× TBST (0.1% Tween-20 in 1 × TBS), it was probed with alkaline phosphatase–conjugated secondary antibody (1:10,000 dilution; Abcam). The blot was developed using AP developing solution (B genie) and the image was captured. The densitometric analysis was performed with the ImageJ software, NIH.
In vitro scratch assay
MiR-21 is known to promote proliferation and migration in tumor cells. To check the antagonizing miR-21 function of various AMOs and how this inhibits migration, we performed scratch repair assay [28]. For this, MCF-7 and MDA-MB-231 cells (3 × 105 cells) were grown to 70% confluence in a six-well plate and transfected with 50 nM of Scrambled, PS modified, AMO-1, and AMO-2. Next, the cells were allowed to attain 90% confluency so that a monolayer is formed. The media was removed after 24 h, and cells were scratched horizontally in the middle of the plate using a sterile 200 μL pipette tip. The debris was washed with 1× PBS and fresh media was replenished. At this time (0 h), the images were taken and cells were placed back in the humidified incubator to allow cell to repair the scratch. Post 24 h, cell images for each treatment were taken and the migration was recorded using Zeiss Microscope. The area of scratch closure was calculated using NIH ImageJ software by the following formula:
where areat =0 h is the area of scratch measured immediately after scratching, and areat=24 h is the area of scratch measured 24 h after performing the scratch.
Cell viability assay
MCF-7 cells were seeded in 96-well plates (6,000 cells/well) and grown to obtain 60%–70% confluency. The following day, cells were transfected with Scrambled/PS/AMO-1/AMO-2 at a concentration ranging from 10 to 100 nM. After 24 h of growth, cells were treated with 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich Co.) and incubated for 3 h at 37°C in CO2 incubator. Subsequently, the medium was decanted and formazan crystals were dissolved in 200 μL DMSO solution. Sample absorbance was measured at 570 nm with a reference wavelength of 630 nm. Data were normalized with the untreated control set.
Assessing intracellular stability
MCF-7 cells were seeded in 12-well plates (50,000 cells/well) and grown till 70% confluency overnight. The cells were lipofected with PS-modified 2′-O-Me RNA, AMO-1, and AMO-2 at 50 nM. Four hours after lipofection, cells were washed and OPTIMEM media was replaced by full DMEM. Cells were harvested at that time (t = 0 h) and RNA was isolated. Similarly, RNA was isolated at 12 and 24 h post-transfection. cDNA was prepared using 1,000 ng of RNA by QuantiMir Kit, SBI. The intracellular levels of transfected oligonucleotides were measured by quantitative real time PCR (qPCR) performed on Roche LightCycler LC480. The forward primer for detection of anti-miR-21 was: 5′ ACACTCCAGCTGGG
Statistical analysis
In all the tissue culture experiments, the plotted data represent the mean from three independent experiments prepared from different biological replicates. The error bars represent standard error. Differences between scrambled/untreated and transfected groups were calculated using unpaired two-tailed Student's t-test.
Results and Discussions
MiR-21 is an oncogenic miRNA that is significantly overexpressed in many tumors and is functionally associated with cellular proliferation, survival, and migration [30]. The MCF-7 cell line was taken as our experimental system as it expresses high endogenous levels of miR-21 [31]. For achieving functional readout of miR-21 inhibition, we used a dual luciferase screen. The reporter plasmid (psiCHECK-2 Vector; Promega) expresses the renilla gene fused with the miR-21 target sites and a firefly gene for normalization of signal accounting for variability in transfection (internal control). We compared the antisense potency of 2′-O-methyl RNA (AMO-1) and the serinyl capped and UR-AMP interspersed 2′-O-methyl RNA (AMO-2) for their ability to suppress miR-21 levels and hence upregulate complementary target levels. We compared the potency of each AMO with respect to chemistry matched 2′-O-methyl scramble RNA to avoid variability caused by transfection itself. Furthermore, we also compared the AMOs to another 2′-O-methyl RNA with full PS-modified backbone, a first generation antisense oligonucleotide known to have nuclease resistance and high binding affinity toward target RNA [22]. The cells were transfected with reporter construct with varying concentrations of the AMOs (10–50 nM) and assayed for renilla and firefly luciferase intensity 24 h post-transfection. Transfection of the oligonucleotides resulted in antagonism of miR-21, as shown by specific increase in the levels of the relative target luciferase intensity (Fig. 2).
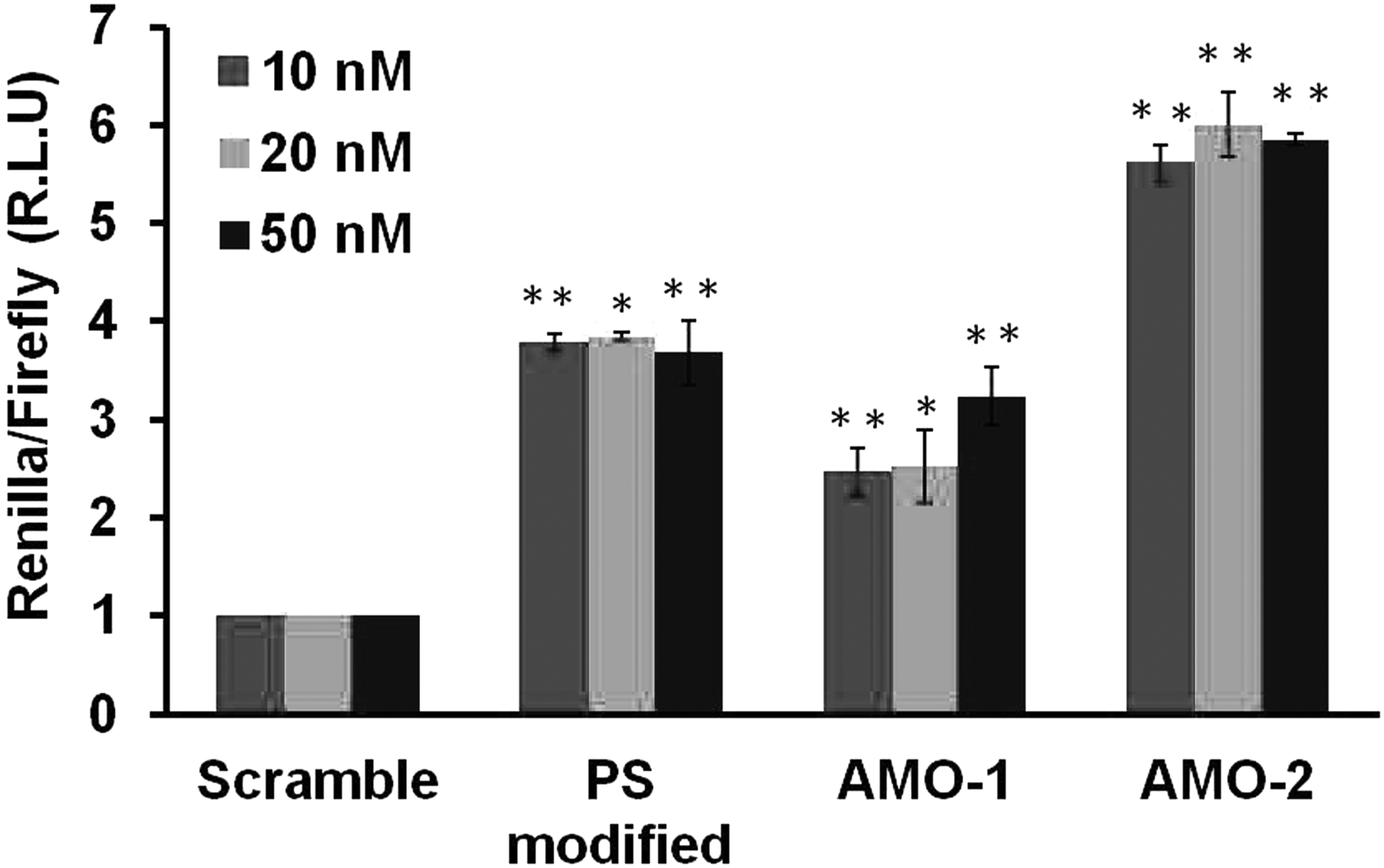
Comparison of the functional potency of antisense treatments against miR-21. Cells were transfected with different concentrations (10–50 nM) of scrambled control, PS modified, AMO-1, and AMO-2 for 24 h. The cell lysates were subsequently analyzed for luciferase signal of target compared to scrambled control. An increase in luciferase signal is observed for different oligonucleotides till 50 nM. AMO-2 shows a maximal increase in luciferase luminescence at 50 nM compared to AMO-1 and PS modified. Data shown are the mean ± SE of three independent experiments. *P < 0.01 and **P < 0.001 indicate significant statistical difference between the oligonucleotide transfected and scrambled control. All wells were normalized with the internal firefly control. PS, phosphorothioates; SE, standard error.
As evident from Fig. 2, an increase in firefly luciferase signal was observed upon transfection with AMOs at all concentrations. AMO-2 showed ∼6-fold increment of luciferase intensity at 50 nM concentrations with respect to 2′-O-methyl RNA scrambled control. PS-modified 2′-O-methyl RNA also showed an efficient derepression of miR-21. Among the 2′-O-methyl RNAs, AMO-2 was found to be the most potent in inhibiting miR-21 levels in all the tested concentrations. These AMOs were found to be functionally potent at 50 nM, thus affirming this concentration to be used for further experiments.
To assess the efficiency of AMO-1 and AMO-2 to inhibit miR-21, we checked the levels of mature miRNA by qRT-PCR 24 h post-transfection with 50 nM and compared the outcome with that of chemistry-matched scrambled control (Fig. 3). The ability to downregulate miR-21 was supreme for AMO-2 with significant decrease of mature miR-21 levels up to ∼95%. AMO-1 and PS-modified AMO showed ∼70% knockdown post-transfection for 24 h compared to scrambled control. AMO-2 was found to efficaciously repress miR-21 levels to a greater extent than AMO-1, which might be due to the fine tuning between high duplex stability and the stability to nucleases conferred by the interspersed UR-AMP and the capping by serinyl units. The AMO-mediated repression was also measured after 48 h (Supplementary Fig. S4) showing ∼71% reduction when transfected with PS-modified AMO, ∼85% by AMO-1, and ∼92% by AMO-2, implicating sustained inhibition of miR-21 levels.
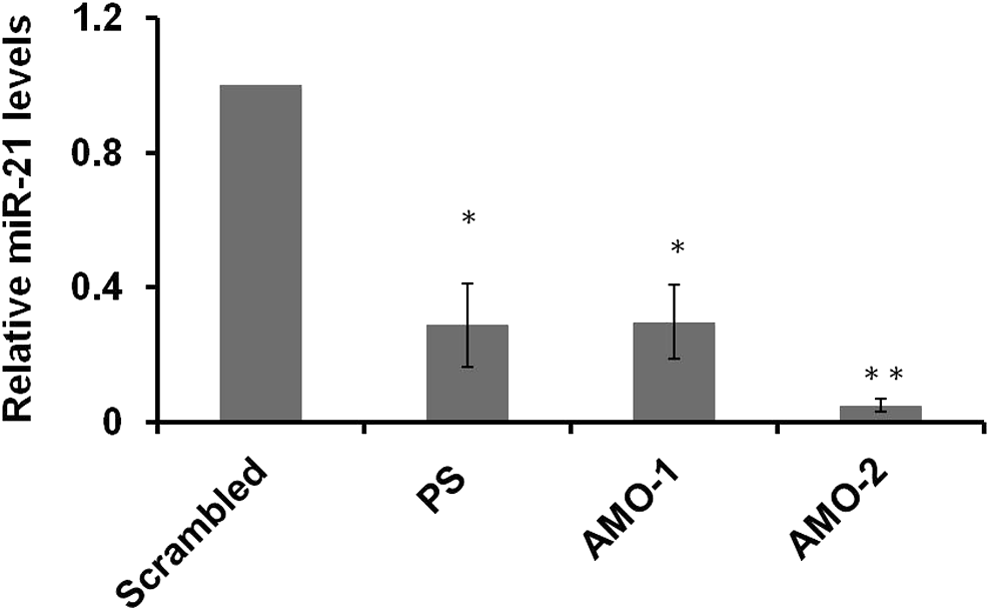
Reduction of miR-21 levels by various antisense oligonucleotides. AMOs designed against miR-21 were transfected at 50 nM concentration, and mature miRNA levels were measured by qRT-PCR after 24 h. Approximately, 95% inhibition of miR-21 is caused by AMO-2 compared to PS modified and AMO-1, which cause ∼70% downregulation. Data shown are the mean ± SE of three independent experiments with *P < 0.01 and **P < 0.0001 (Student's t-test). qRT-PCR, quantitative real-time reverse transcriptase–polymerase chain reaction.
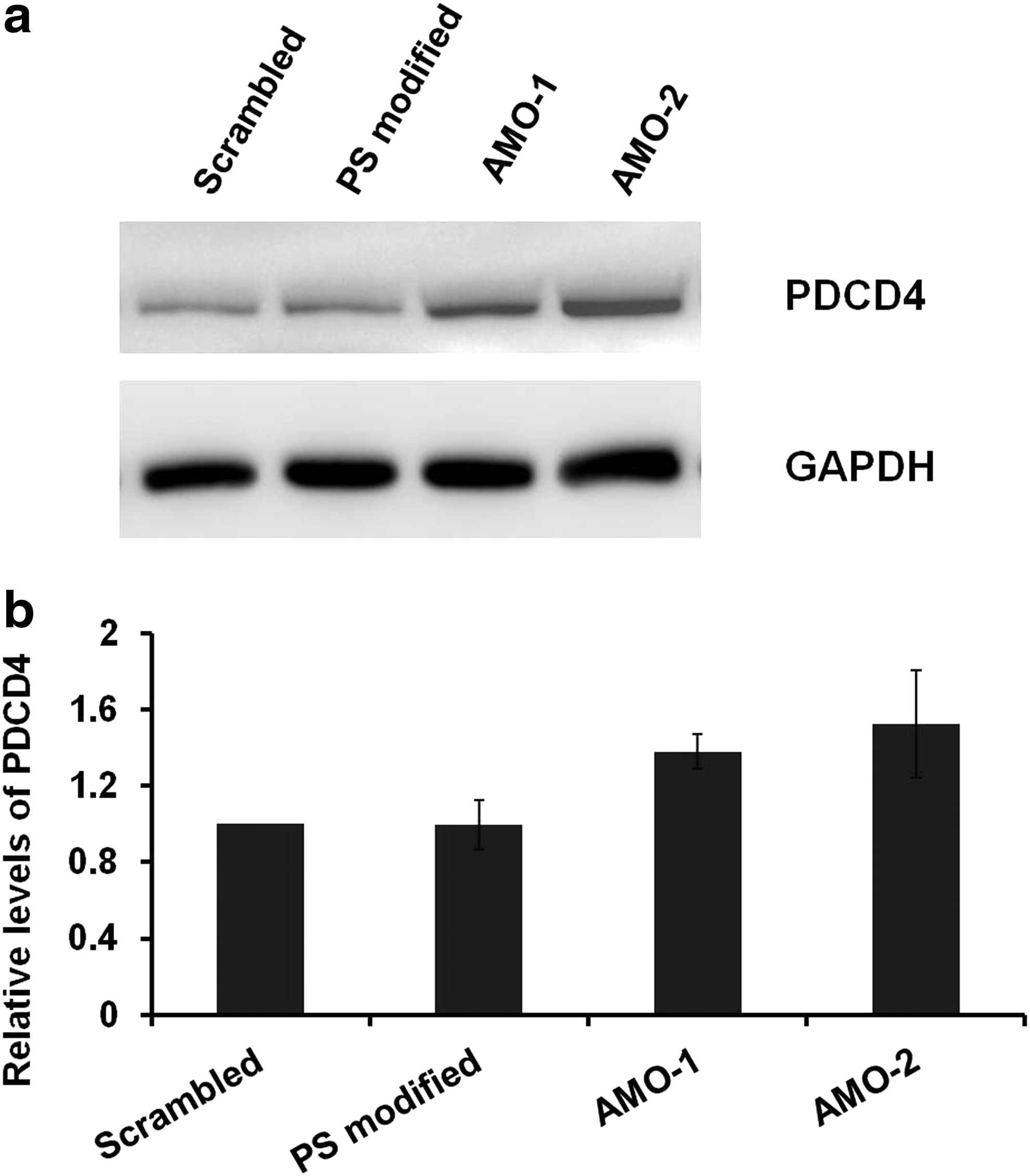
Translational repression is considered as a major mechanism for miRNA regulation of target gene expression. Tumor suppressor, PTEN, is a principle target of miR-21 [32]. PDCD4 is an important validated target of miR-21, which is downregulated by miR-21 in many cancers [33]. Overexpression of miR-21 has an inverse correlation with its target PDCD4 and PTEN protein levels. Thus, the extent of suppression of miR-21 targets, PDCD4 and PTEN, was examined by western blot (Fig. 4 and Supplementary Fig. S5) to evaluate the antisense potency of the AMOs after 24 h of treatment. It was shown that the PDCD4 and PTEN levels were increased after transfection with modified AMOs compared to 2′-O-methyl scrambled control. The target levels were upregulated to a greater extent when treated with AMO-2 (with a P value <0.05) compared to scrambled control than AMO-1. However, the effects of derepression of targets were typically modest consistent with findings of recent high-throughput proteomic approaches [34,35]. Surprisingly, PS-modified RNA did not have an effect on the endogenous protein levels post 24 h.
Tumor cell invasion is vital step for cancer cells to metastasize, and miR-21 is known to promote migration and proliferation in cancer cells [36]. We sought to envisage the extent to which knockdown of miR-21 by various AMOs would arrest the capacity of cancer cells to metastasize. For this, we took two different cell lines differing in their properties to metastasize, MCF-7 and MDA-MB-231. MDA-MB-231 has proved to be a highly metastatic model to study migration properties [37] and expresses high levels of miR-21 [36]. To establish that knockdown of miR-21 antagonizes its activity, we transfected each of the chemically modified RNAs and performed a scratch assay (Fig. 5 and Supplementary Figs. S6 and S7). After 24 h, we observed that the cells treated with AMOs had delayed migration compared to the untransfected cells and scramble transfected cells. Knockdown of miR-21 suppressed migration capacity by ∼19.4-fold in PS-modified AMO, ∼20-fold of AMO-1, and ∼33.4-fold in case of AMO-2 in MCF-7 cells compared to scrambled control. In MDA-MB-231, the suppression was ∼17.1-fold in PS modified, ∼16.2-fold in AMO-1, and ∼21.2-fold in case of AMO-2 compared to scrambled control. The difference between scrambled control versus PS modified and AMO-1 had a P value <0.05, whereas the difference between scramble treated and AM0-2 had a P value <0.001.
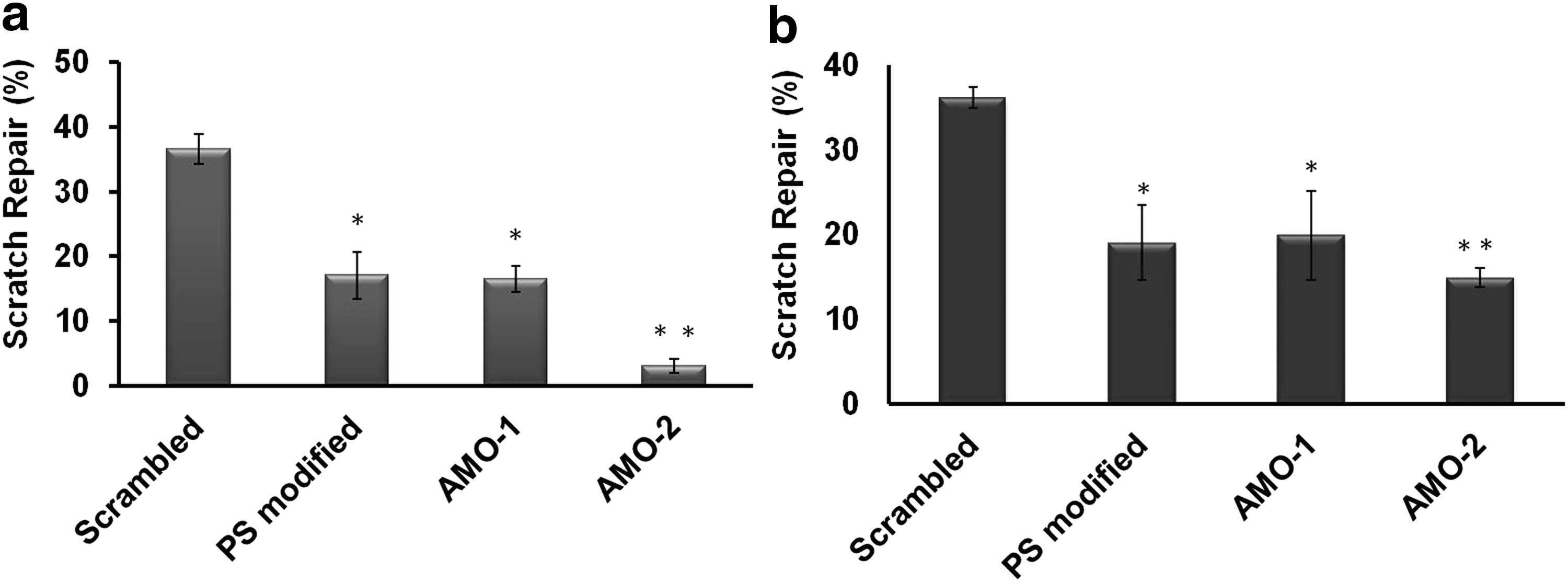
In vitro scratch assay in
The challenge of cytotoxicity has always remained an important point of consideration for chemically modified oligonucleotides for their use for therapeutic intervention. To examine this, we performed concentration-dependent cell viability assay from a range of 10 to 100 nM. We observed that there was no measurable cytotoxicity for AMO-1, AMO-2 even at 100 nM rendering them as suitable candidates for the development of potential therapeutics in future (Supplementary Fig. S8). However, PS-modified AMO showed significant toxicity up to ∼40% at 100 nM. This observation was consistent with previous studies that implicated cytotoxicity as one of the potential limitations of PS backbone [38,39].
Furthermore, we assessed the relative intracellular stability of these chemically modified AMOs by qPCR method. A comprehensive analysis of detection and quantification of modified siRNA using real-time polymerase reaction has been shown previously. Although the modified oligonucleotides are detected at lower efficiency, it does not limit the utility of the assay [40]. To determine whether reverse transcriptase is able to use modified oligonucleotide as template for cDNA synthesis, we polyadenylated all the different oligonucleotides, followed by oligo (dT) hybridization and reverse transcription to cDNA. We then compared the reverse transcription profiles obtained from in vitro synthesis of cDNA for each AMO. The standard curves were plotted for each AMO with Ct (threshold cycle) values versus concentrations of cDNA synthesized (Supplementary Fig S9a). The qPCR efficiency was determined from the slope of the log plot of concentration versus Ct value. The calculation for conversion of slope to efficiency was done using the formula:
By looking at the standard curve and slopes for each AMO, we found that AMO-1 and PS modifications were detected efficiently compared to AMO-2, which was detected although with poor yields. The serinyl capping present in AMO-2 is a non-nucleotide modification, but contains a free OH group at the 3′ end, which can be utilized for polyadenylation. Thus, we find amplification after qRT-PCR for AMO-2 with lesser yields compared to uncapped AMO-1 and PS-modified oligonucleotides (as confirmed by the Ct values), but this did not limit the utility of the assay to detect their intracellular levels. We also monitored the exponential amplification for the oligonucleotides following conversion to cDNA with antisense-specific forward primer and oligo (dT)-specific reverse primer by gel electrophoresis. As a negative control, we included an unmodified RNA without poly (A) tailing subjected to cDNA synthesis using same conditions. The PCR products were observed as a single band (Supplementary Fig. S9b). Once the assay was established, we transfected the oligonucleotides at 50 nM and measured their relative levels at 0, 12, and 24 h by qRT-PCR (Supplementary Fig. S9c). Post 24 h, AMO-2 was found to be most stable (∼68%) compared to AMO-1 (∼22%) and PS modified (∼38%) owing to the UR-AMP-interspersed serinol capping. However, the transfection of AMOs did not alter the miR-21-3p (star strand) levels after 24 h (Supplementary Fig. S9d).
In summary, we have successfully designed and evaluated the effectiveness and antisense potency of uniformly substituted 2′-O-methyl RNA (AMO-1) and UR-AMP-interspersed serinol capped 2′-O-methyl RNA (AMO-2) against miR-21. We demonstrate that the inhibitory efficacy of full 2′-O-methyl RNA is enhanced by serinyl capping at 5′/3′ ends and 2′-O-(R-2-amino-3-methoxypropyl) (2′-R-AMP) modification of uridine at three distinct positions. These modifications render the AMO-2 resistant to nucleases present in the cell thereby enhancing its intracellular stability while preserving the duplex stability, making it a more effective inhibitor of miR-21. There can be multiple mechanisms of miRNA inhibition, in response to transfection of AMOs [10]. The AMOs primarily act by sterically blocking the mature miRNA from binding to its target mRNA [41]. The other mechanisms include recruiting and triggering RNase H-dependent degradation of miRNA [42], or sequestering miRNA in P-bodies followed by its decay [14,43,44]. In comparing different chemistries of PNA-K3, LNA/DNA, or LNA/2′-OMe mixmer, Torres et al., observed that only 2′-O-methyl AMO was able to cause degradation of miR-122 in Huh cells, whereas high affinity K-PNA-K3, LNA/DNA, or LNA/2′-OMe mixmer AMOs did not lead to degradation of the target miRNA when bound in miRISC [14,43,44].
Conclusions
miRNAs are small noncoding RNAs that accomplish crucial roles in major biological processes. The development of anti-miR oligonucleotides as tools for deciphering miRNA functions or as probes for miRNA detection or as therapeutic modules necessitates the identification of optimal chemical modifications to endow better nuclease resistance, as well as enhancing potency. This novel 2′-R-AMP modification in uniformly 2′-O-methyl RNA with serinyl end caps is a simple and novel strategy that fine tunes the balance between binding affinity and nuclease resistance leading to highly potent inhibitor of miR-21, with low toxicity.
Footnotes
Acknowledgments
The authors acknowledge the support by Council of Scientific and Industrial Research (CSIR) (project title: Genome Dynamics in Cellular Organization, Differentiation and Enantiostasis project code BSC0123), India. The authors thank Dr. Hemant Suryawanshi for providing the miR-21 target luciferase construct.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.