
Abstract
This review summarizes the current understanding of nephrotoxicity related to the administration of therapeutic oligonucleotides, particularly those with 2′-methoxy-ethyl (2′-MOE) modifications. To best understand the effects of antisense oligonucleotides (ASOs) on the kidney, the reader should have a general understanding of renal microanatomy, physiology, and general mechanisms related to toxicity, so a short review is presented. Preclinical–clinical correlates are also discussed. Collectively, the data for PS ODN and 2′-MOE-modified ASOs have shown the laboratory animal species utilized in toxicology studies generally overpredict renal effects of these agents. As such, 2′-MOE ASOs do not appear to pose as much of a risk to patients as the preclinical data would suggest. This observation has been confirmed so far in clinical investigations.
Introduction
A
It is well known that the kidney is the primary site of oligonucleotide distribution following parenteral administration, achieving higher concentrations than any other organ and accounting for up to 20% of the total administered dose [2]. The systemic effects of ASOs have been widely studied and target organ toxicities have been described with the kidney as a primary target organ in rodents and nonhuman primates [3]. ASOs are readily filtered through the glomerulus and rapidly reabsorbed by the proximal tubule epithelium [3]. The primary focus of this review is the comparative effects of single-stranded ASOs on renal structure and function, but one needs to understand the functional anatomy and physiology of the kidney to fully appreciate the potential effects, or lack thereof, of ASOs on the kidney. Therefore, a concise review of renal anatomy and physiology and renal toxicology is presented first.
General Concepts in Renal Toxicopathology
The kidneys are exceptionally vulnerable to toxic injury for several reasons. Although the kidneys constitute less than 1% of body weight, the renal blood flow accounts for 20% of the cardiac output, and 90% of the blood entering the kidneys goes to the renal cortex. When the extensive blood flow is coupled with the glomerular capillary system, the result is an extremely large potential for the presentation of toxic substances to a vast endothelial surface area in the renal cortex. In addition, as the kidney consumes oxygen at an extraordinary rate, it is very susceptible to substances that cause cellular hypoxia. Furthermore, the countercurrent mechanism of the kidney predisposes the medullary interstitium to progressively increasing concentrations of certain nephrotoxic substances, such as heavy metals and β-lactam antimicrobials [4].
The cortex and medulla each have distinct biochemical activities, which function in the metabolism of pharmacologic agents and xenobiotics [5]. The kidneys possess the necessary drug-metabolizing enzymes that not only detoxify chemicals but also may create metabolites that are more toxic. These actions combine to increase the susceptibility of the kidney to chemically induced damage. The adverse reaction may be in response to the parent compound, toxic metabolites, or both. Tubular secretion and reabsorption increase the tubular concentration of these compounds to concentrations that are toxic to renal tubular epithelium, although the remainder of the body may be subjected to far lower concentrations [6].
Anatomy and Physiology of the Glomerulus
The normal glomerulus, an exquisitely adapted dynamic sieve, forms an ultrafiltrate that is virtually devoid of blood cells and plasma proteins. The ultrafiltration membrane includes the fenestrated capillary endothelium, the glomerular basement membrane, and the visceral epithelial cells or podocytes. The entire glomerulus is enmeshed with mesangial cells and their accompanying intercellular matrix (Fig. 1). Chemical-induced changes in these structures, either by direct action or immune mediated, affect several physiologic parameters that govern the single-nephron glomerular filtration rate (GFR), such as transcapillary hydraulic pressure, glomerular plasma flow rate, glomerular filtration surface area, and the intrinsic permeability of the glomerular filtration barrier [7].
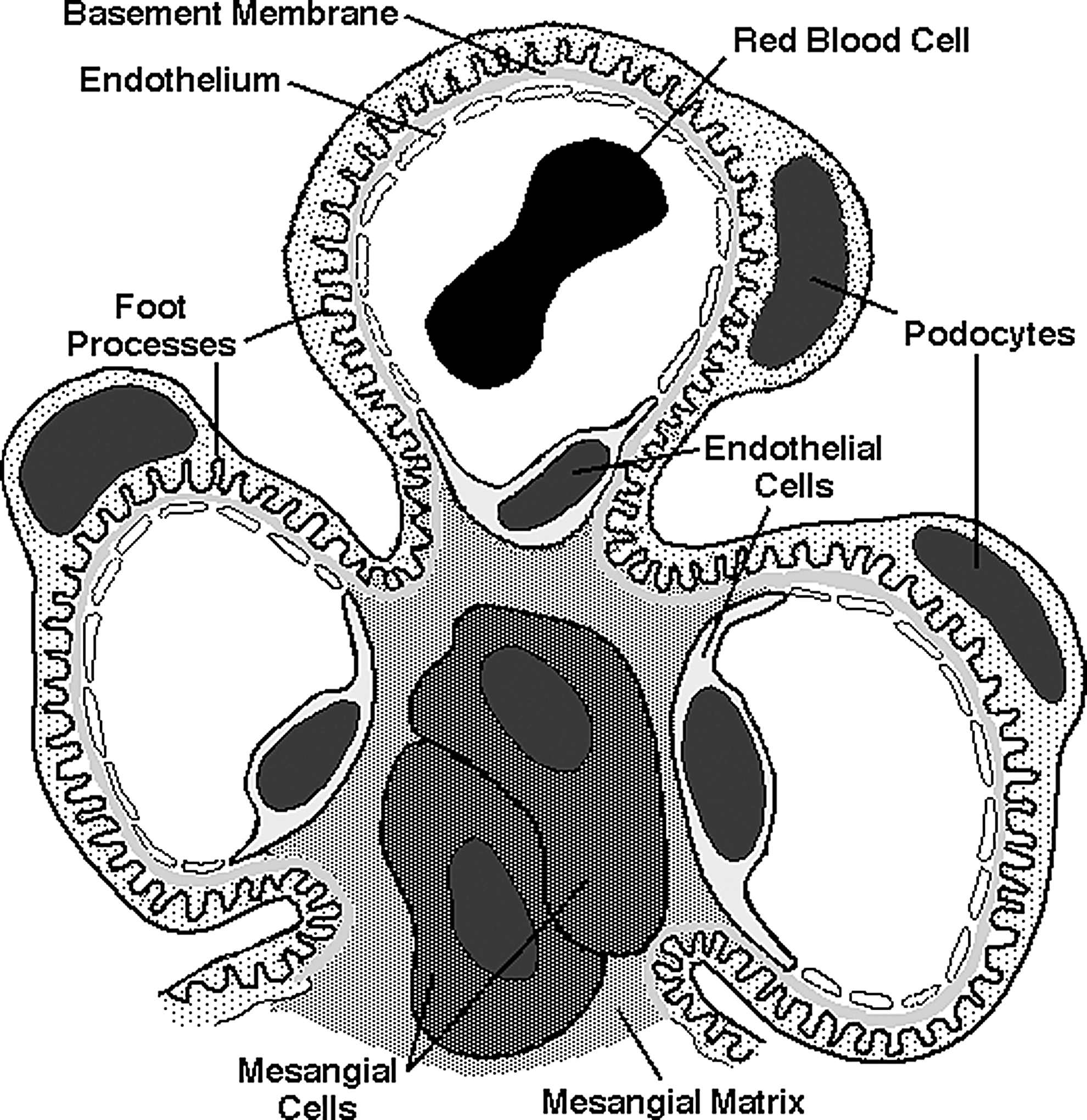
Schematic architecture of the glomerulus showing podocytes, podocyte foot processes, mesangial cells, mesangial matrix, endothelial cells with endothelial pores, glomerular basement membrane, and a red blood cell within a glomerular capillary.
The glomerular capillaries and mesangium determine the functional surface area for ultrafiltration. Alteration of a luminal factor at the loop of Henle is detected by the macula densa at the distal tubule. The exact luminal factor has not been precisely determined. Increased perfusion at the loop of Henle causes an increase in the intraluminal concentration of sodium, and GFR decreases. The decreased GFR may be the result of a change in the salt concentration of the tubular fluid rather than in the flow rate. This change has correlated best with chloride ion concentrations as blockade of chloride excretion by furosemide significantly decreases the GFR response [8]. The macula densa releases renin, which then activates angiotensin II [9]. Angiotensin II can constrict the afferent renal arterioles to decrease glomerular plasma flow. Angiotensin II also constricts the mesangium to decrease the filtration surface area of the glomerulus. Both of these changes effectively decrease GFR [8].
Therefore, any drug that either affects the renin–angiotensin system (e.g., captopril) or alters renal blood flow (e.g., epinephrine) can alter the glomerular ultrastructure and, thus, the filtration surface area. Because of this tubuloglomerular feedback, tubular toxins can also produce dose-related glomerular alterations [10]. Specifically, aminoglycosides decrease both the diameter and the density of the endothelial fenestrae, thereby decreasing the surface area for ultrafiltration [11]. Other tubular toxins may also produce similar glomerular changes.
Mesangial cells are also phagocytic, being derived from the monocyte–macrophage system. Administration of cytostatic drugs, such as azathioprine, penicillamine, and daunorubicin, results in increased amounts of mesangial matrix and proliferation of mesangial cells. These drugs may alter mesangial cell function, thereby allowing accumulation in the matrix of macromolecules that would normally be phagocytized by mesangial cells. As a result, feedback recruitment of additional phagocytic cells may occur, producing the histologic appearance of membranous glomerulonephritis.
Mechanisms of Glomerular Damage
The plasma membranes of visceral epithelial cells are rich in anionic sialoglycoproteins, which not only act as an electrostatic barrier against negatively charged molecules but also serve to maintain normal separation of podocyte foot processes [12]. When highly cationic molecules disrupt the negative charge of the podocytes, the podocytes become swollen, foot processes become blunted, cell junctions between foot processes are formed, and the slit diaphragms between adjacent foot processes are destroyed. Functionally, the barrier against negatively charged macromolecules, such as albumin, is disrupted and the selectivity of the ultrafiltration apparatus diminishes [13]. Decreases in the fixed negative charge of the visceral epithelial cells also cause accumulation of macromolecules in the mesangial matrix [14]. These electrostatic imbalances can be reversed by perfusion with heparin, a negatively charged glycoprotein [12]. Drugs that cause loss of these anionic glycoproteins include puromycin [13], daunorubicin [14], adriamycin [15], and probenecid [16]. Other organic acids may cause similar glomerular changes.
Most drug- and chemical-related glomerulonephropathies are immune mediated. The drug may act as a hapten or as a full antigen to cause type III or, less frequently, type II hypersensitivity. On the other hand, some drugs cause a systemic lupus erythematosus (SLE)-like syndrome, in which case, antinuclear or anti-DNA antibodies are elicited [17]. In these situations, immune complexes are deposited in the subepithelial layer, the glomerular basement membrane, or the mesangial matrix. The subsequent activation of complement leads to the liberation of cytotoxic free oxygen radicals from cells of the monocyte–macrophage system and recruitment other inflammatory components [18]. Drugs that have been shown to cause deposition of immune complexes include penicillin [19], penicillamine [20], and gold salts [21]. Drugs that produce SLE-like reactions in humans include procainamide, probenecid, hydralazine, and isoniazid.
Anatomy and Physiology of the Tubular Network
The renal tubular network of the nephron is composed of the proximal tubule, loop of Henle, distal tubule, and collecting duct (Fig. 2). The morphology, metabolic activity, and function of the tubular epithelial cells vary from the proximal tubule to the collecting duct, as does their susceptibility to nephrotoxic compounds.
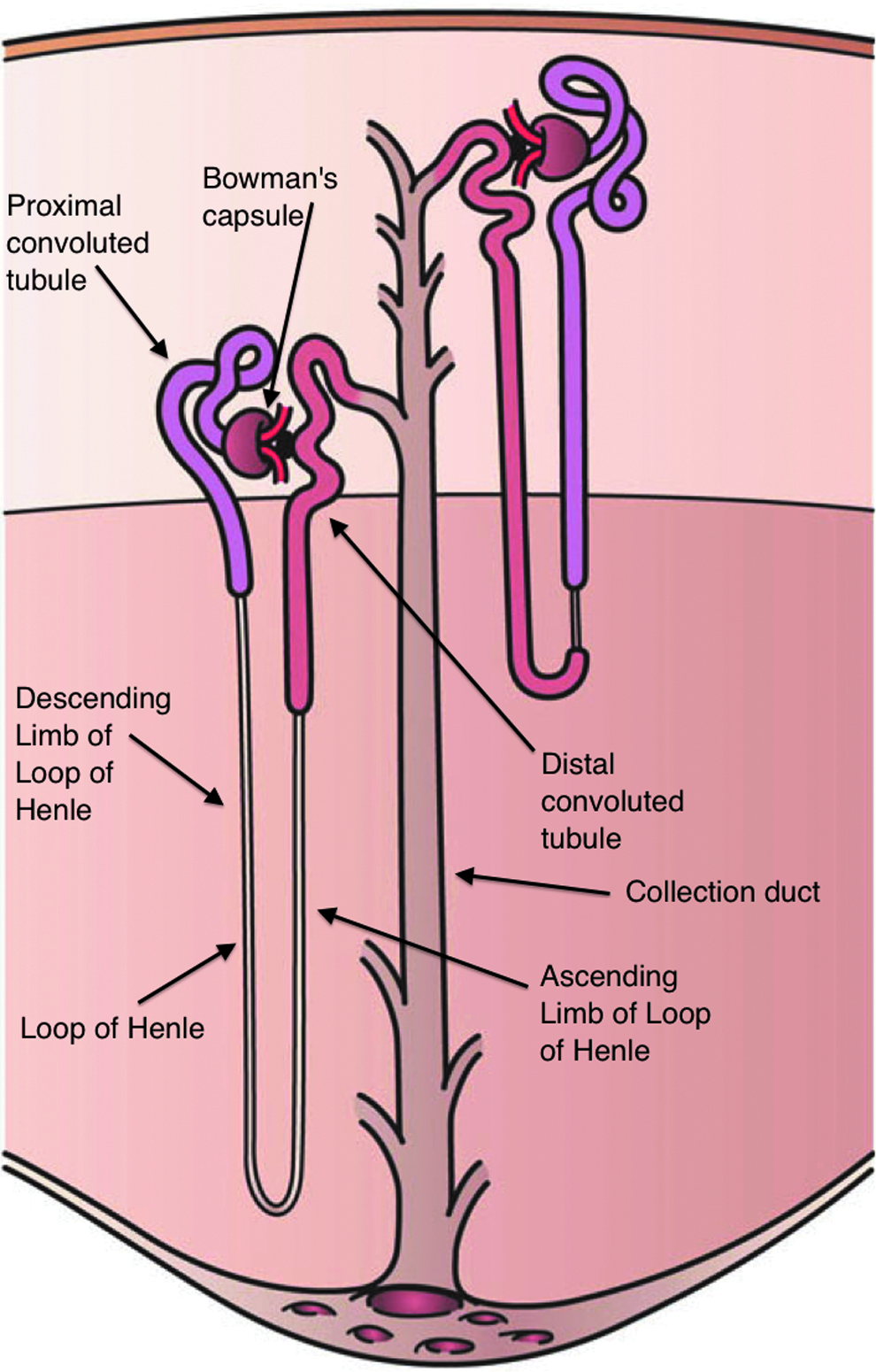
Schematic organization of the tubular network showing the cortex and medulla and depicting Bowman's capsule (glomerulus), proximal convoluted tubule, the loops of Henle, the distal convoluted tubule, and a collecting duct. The glomeruli are located in the cortical region, whereas tubules are present in both the cortex and medulla.
Proximal tubules
The proximal tubule begins at the urinary pole of the renal corpuscle, with the tubular epithelium continuous with the epithelium of Bowman's capsule. The proximal tubule initially has a convoluted structure (pars convoluta; S1) and then assumes a straight path (pars recta; S2/S3) as it continues. A single layer of cuboidal to columnar epithelial cells lines the proximal tubule. Numerous mitochondria and extensive rough and smooth endoplasmic reticulum impart a high degree of metabolic activity to these cells. The luminal border is lined by well-developed microvilli and an extensive membrane-bound enzyme system, which includes alkaline phosphatase, gamma-glutamyl transaminase, and β-N-acetylglucosaminidase. Adenosine triphosphatases (ATPases) are present on both the luminal and basolateral membranes, but predominantly on the latter [22–24].
Functionally, the proximal tubule is responsible for the active reabsorption of glucose, protein, amino acids, nucleic acids (including ASOs), sodium, potassium, calcium, phosphate, and bicarbonate. In addition, chloride, water, and urea are passively resorbed, and hydrogen ion is actively secreted [4,8].
Loops of Henle
The loops of Henle are continuous with the proximal tubule and are lined by a single layer of low cuboidal to squamous epithelium. The length of the loop is related to the position of the nephron within the kidney, with longer loops in juxtamedullary nephrons and shorter loops in cortical nephrons [23]. The metabolic activity of the epithelial cells is much less than in other tubular segments. The loops of Henle function primarily to generate a gradient of solute concentration within the medullary interstitium to concentrate urine [4].
Distal segments
The distal tubule is composed of the straight portion (pars recta); the portion adjacent to the renal corpuscle, containing the macula densa (pars maculata); and the convoluted portion (pars convoluta). The single layer of tubular epithelial cells is cuboidal to columnar, but generally lacks apical microvilli. Mitochondria and other subcellular organelles are less abundant than in the proximal tubule [23].
Functionally, the distal tubules actively reabsorb sodium, calcium, and bicarbonate and passively resorb chloride and water. Hydrogen ions, ammonium ions, and urea are actively secreted, whereas potassium is passively secreted [4].
Collecting ducts
The collecting ducts, which complete the renal tubular network, are lined by cuboidal to columnar epithelial cells. As the ducts progress distally, the epithelial height increases. Mitochondria are present in approximately the same concentration as in the distal tubule, supplying the energy necessary for active reabsorption of sodium and active secretion of hydrogen ion. Chloride and water are passively resorbed and potassium passively secreted [4].
Mechanisms of Tubular Damage
As mentioned above, the morphology, metabolic activity, and innate functions of the tubular epithelial cells make them quite susceptible to xenobiotic injury. This is particularly true for the proximal tubule. The response to a particular injury varies with the severity of the insult. If an agent is not directly toxic to the tubular epithelium, the mechanisms of nephrotoxic tubular damage are similar to those of ischemic tubular necrosis [14]. Several theories have been presented to explain tubular necrosis. Historically, passive diffusion, leakage of tubular fluid into the interstitium, or mechanical obstruction due to the presence of intraluminal casts was considered the principal mechanism of tubular damage. These theories of back-leak and tubular obstruction were based on morphologic studies in which intraluminal casts were consistently found in association with tubular necrosis and basement membrane disruption. Alterations in GFR could not be explained by obstruction or backflow of filtrate, however, and these theories are no longer widely accepted [25].
One current theory suggests that the initial nephrotoxic or ischemic insult produces hypoxic endothelial cell swelling in renal arterioles, apparently through ischemic impairment of the sodium pump. The endothelial swelling then leads to a decrease in glomerular permeability [26]. This theory has not been proven in the clinical setting.
The factor most widely recognized as the cause of nephrotoxic or ischemic alterations is the vasoconstriction that results from persistent vasomotor hyperactivity. The renin–angiotensin system may play an important role in this mechanism and is discussed further as it relates to renal papillary necrosis and interstitial toxicity. The resultant profound vasoconstriction decreases renal blood flow and, thus, GFR [27].
Chemicals are often directly toxic to renal tubular epithelium. Because of the heterogeneity of cell types present within the renal tubules, the tubules have the capacity to perform many complex and diverse functions simultaneously. This high level of biochemical activity also makes the proximal tubules the portion of the nephron that is most susceptible to toxic compounds. Androgens also appear to play a role in tubular susceptibility to various nephrotoxins [6].
For most nephrotoxicants, specific mechanisms of cellular or subcellular damage are not understood, but there are several possibilities. Compounds may directly affect tubular function by multiple mechanisms, including interference with tubular membrane transport systems, alterations affecting subcellular metabolism, and finally damage to specific organelles [28].
The mixed-function oxidase system is principally responsible for the oxidative biotransformation of many endogenous and exogenous compounds [5,29]. Cytochrome P450 is a unique hemoprotein that functions as the terminal oxidase for the mixed-function oxidase system [30]. The distribution of this system within the kidney varies among species [28], but the highest concentrations tend to be found in the pars recta of the proximal tubule [31].
Membrane effects
Compounds that induce nephrotoxicity by altering the cell membrane do so by surface-active properties or interfering with ATP-mediated transport activities. Polymyxins and amphotericin B exert their effects, in part, by disrupting the normal membrane sterol–lipid interactions, allowing the membranes to become more permeable to hydrogen and other ions [6]. Breakdown of the permeability barrier leads to osmotic disruption of the cell. Gentamicin and other aminoglycosides tend to affect the membrane by interfering with the Na+-K+-ATPase system [32]. This disruption, in turn, alters organic ion and bicarbonate transport, leading to tubular dysfunction [33]. Similar effects are produced by cephaloridine and cephalothin [34]. Cephaloridine also causes peroxidation of membrane lipids [35], which interfere with ion transport and depletion of glutathione that contributes to membrane and subcellular alterations [36]. Membrane effects tend to be most marked in the proximal tubule [37]. Sulfonamides that precipitate in the tubules bind to membrane proteins and interfere with ion transport and osmotic balance [38].
Effects on subcellular metabolism
The creation of metabolites that are capable of covalent binding with cellular macromolecules represents another general nephrotoxic mechanism [39]. Metabolites of acetaminophen form covalent links with endoplasmic reticulum, leading to cell dysfunction through interruption of normal protein synthesis [40]. Out-dated tetracycline contains degradation products that may produce a reversible Fanconi-like syndrome (glycosuria, aminoaciduria, and hyperphosphatemia), which is most likely related to the accumulation of metabolites within mitochondria resulting in the disruption of cristae and inhibition of many oxidative enzymes involving glycolysis, the Krebs cycle, the hexose monophosphate shunt, and β-oxidation of fatty acids. This effect, seen solely in the proximal tubule, leads to progressive nephropathy. In normal clinical use, tetracycline only causes slight functional impairment [6]. Interruption of the energy-producing machinery of the tubular cells begins the potential cascade leading to cell death and, eventually, disruption of tubular function.
Damage to specific cytoplasmic organelles
An early event in aminoglycoside nephrotoxicity is the accumulation of phospholipids and antibiotics within the lysosomes of proximal tubular epithelial cells. The accumulation of phospholipids results from aminoglycoside inhibition of phospholipases A and C [41] that leads to renal phospholipidosis [42], a proposed mechanism for the formation of intracellular myeloid figures or cytosegresomes [43]. The enzyme inhibition apparently results from binding of the aminoglycosides to phospholipids (e.g., phosphatidylinositol) and alteration of the substrate for phospholipases [44]. The physiologic significance of phospholipase inhibition remains unknown, but may be related either to the role of these enzymes in prostaglandin synthesis and degradation [41] or to the induction of lysosomal instability [42].
Mechanisms of Damage to the Renal Papilla and Interstitium
The pathophysiology of chemical-related papillary and interstitial disease involves either hypersensitivity or inhibition of protective prostaglandins by nonsteroidal anti-inflammatory drugs (NSAIDs). The hypersensitivity reactions generally arise from the penicillin class of antibiotics. Interstitial nephritis develops in only a small percentage of patients receiving penicillin or its derivatives and the accompanying clinical signs are consistent with an immune mechanism. Clinical indices often include a high IgM titer and IgG antibodies directed against the penicilloyl moiety. Cell-mediated immunity may also be operative in drug-related interstitial nephritis. The penicilloyl haptens may uniquely bind to renal proteins to form hapten-renal protein conjugates [45].
Renal papillary necrosis resulting from medullary ischemia is most likely caused by inhibition of prostaglandin synthetase; the decrease in enzyme activity causes a decrease in the production of renal prostaglandins with a concomitant increase in leukotriene production [46]. During systemic vasoconstriction, the formation of angiotensin II causes the localized production of vasodilatory renal prostaglandins to offset the vasoconstrictive action of angiotensin II. NSAIDs decrease renal prostaglandin production, thereby allowing the vasoconstrictive action of angiotensin II to predominate in the renal vasculature. Further vasoconstriction is caused by the increased leukotriene production that results from blockade of cycloxygenase and the consequent increase in the amount of substrate diverted through the lipoxygenase pathway, which is responsible for leukotriene production. The result is profound vasoconstriction, which causes decreased renal blood flow followed by ischemic necrosis of the renal papilla [47,48]. Phenylbutazone, phenacetin, aspirin, acetaminophen, and indomethacin are all capable of causing renal papillary necrosis, apparently by causing medullary ischemia by prostaglandin synthetase inhibition [46].
The development of reliable and reproducible animal models is necessary for the evaluation of nephrotoxic potential of chemicals and pharmaceuticals. Given the structural and functional heterogeneity of mammalian nephrons, not all species, or even all breeds or strains within a species, will have the same degree of nephrotoxic susceptibility to pharmacologic and xenobiotic agents [49]. Furthermore, extrapolation of nephrotoxic potential across species lines is also dependent upon the relative size and metabolic rates of each species.
PS ODN and 2′-Methoxy-Ethyl ASOs and the Kidney
The toxicological profile of first-generation PS ODNs and second-generation 2′-methoxy-ethyl (2′-MOE) ASOs in the kidney are comparable [1]. The only difference is that the stability that leads to the longer clearance half-life for the 2′-MOE ASOs can result in higher kidney concentrations. The kidney contains the highest concentration of oligonucleotide following parenteral administration and is a primary target organ for toxicity in monkeys and rodents. This high concentration of an ASO is due to the physiological handling of this material by the kidney in which the ASO that is filtered through the glomerulus is quickly reabsorbed by the proximal tubular epithelium [50]. ASOs may enter the epithelial cell through the apical brush border or basolateral surfaces, but most appear to be taken by endocytosis at the brush border, much in the same way as these cells reabsorb low-molecular-weight proteins from the urinary filtrate [51]. As such, the uptake and processing of oligonucleotide by these cells is consistent with the functional role that the proximal tubules play in the kidney. The uptake appears to be receptor mediated as other polyanions such as dextran sulfate will compete with ASOs for transport, but the specific receptor(s) remains unknown [52,53].
Immunohistochemistry is the primary method for determination of oligonucleotide concentrations in the kidney and has shown particularly high concentrations localized in the proximal tubular epithelium [50,54]. Following uptake, the majority of oligonucleotides reside in membrane-bound vesicles, such as endosomes and lysosomes, which has been confirmed by ultrastructural analysis [51]. At the higher doses utilized in toxicology studies, proximal tubular epithelial cells can have very high concentrations of ASOs [1,55]. It is these high concentrations that lead to the common histologic alterations in the kidney of cytoplasmic basophilic granules with or without vacuolation of the cytoplasm and are typically limited to the proximal tubular epithelium [56,57]. At sufficiently high doses, degeneration of proximal tubule epithelial cells accompanied by increased secretion of low-molecular-weight proteins can be observed [57].
Uptake of ASOs in rats, mice, and monkeys leads to accumulation of basophilic material in phagolysosomes and is generally dose proportional. Whether or not there is a common endosomal pathway across the species is not clear. Since effects in the kidney are governed by drug concentration, subtle differences in pharmacokinetics related to saturation of plasma protein binding and spilling of drug into the urine may limit tissue exposure in the mouse in addition to a shorter half-life that alter renal accumulation will affect the toxicity profile in the kidney. The relative rate of clearance for a given species will also influence any observed changes, so morphologic alterations are more commonly observed in monkeys relative to mice, where tissue half-lives are shorter.
Effects on the glomerulus
PS ODNs and 2′-MOE ASOs have, so far, not been associated with direct effects on glomerular morphology or function [50,54,58]. In an investigative study in cynomolgus monkeys, histolopathologic, ultrastructural, clinical pathology, and urinalysis evaluation at doses up to 20 mg/kg/week given for 13 weeks or 40 mg/kg/week given for 4 weeks failed to identify any primary toxic effects on the glomerulus [58]. In my personal experience conducting the histopathologic evaluation of repeated dose toxicity studies in mice and primates of 4–39 weeks in duration with more than 60 2′-MOE ASOs (fully modified or mixed backbone), I have not seen primary effects on the glomerular architecture. The only histological effects that I have noted are in primates secondary to activation of the alternate complement pathway cascade with subsequent activation of inflammatory cytokines and chemokines. These events lead to hypertrophy and/or hyperplasia of the mesangial cells of the glomerulus and are not unexpected since these cells are part of the monocyte–macrophage system and the glomerulus is also a source of complement factors. Even when these lesions occur, changes in clinical pathology and urinalysis parameters are typically nil.
Effects on the proximal tubules
As mentioned above, the highest concentration of ASOs in the kidney is localized in the proximal tubule epithelium. The key histologic feature of PS ODNs or 2′-MOE ASO exposure and uptake is the presence of basophilic granules in the cytoplasm of the proximal tubule epithelial cells. Consistent with endogenous nucleic acids, oligonucleotides stain blue with hematoxylin. So, cells that accumulate ASOs or their metabolites, such as the proximal tubular epithelium, will have increased cytoplasmic granular basophilic staining [55–57]. As such, basophilic granules are considered to represent the presence of ASOs and compartmentalization within the cell (Fig. 3). As the distributional pattern of basophilic granules is similar to that for normal low-molecular-weight protein substrates, such as β2-microglobulin, the presence of basophilic granules is not considered to be an adverse toxicologic event in the absence of degenerative changes in cell morphology, but merely represents the cellular uptake of filtered ASOs [3].

Histologic section of kidney cortex from a nonhuman primate given repeated subcutaneous doses of an ASO at 20 mg/kg/week for 13 weeks. Note the presence of abundant basophilic granules within the proximal tubular epithelial cell cytoplasm (arrows). Several tubules have minor vacuolation of epithelial cells at the luminal border. ASO, antisense oligonucleotide.
In addition to the presence of basophilic granules, minimal to moderate degrees of vacuolation of the tubular epithelium cytoplasm is often seen in monkeys given doses >10 mg/kg/week [56]. Vacuolation of these epithelial cells may also occur as a spontaneous background alteration due to the extensive vacuolar (endosomal/lysosomal) apparatus present in these cells. However, the size and incidence of these vacuoles can be significantly increased following treatment with an oligonucleotide.
Vacuolation is generally localized to the primal tubule and is characterized by the presence of single-to-multiple large clear vacuoles within the cytoplasm. In some instances, vacuolation is associated with cellular swelling and what appears to be rupture of the epithelial cells with membrane fragments extending into the tubular lumen. Ultrastructural examination has shown the vacuoles to be distended phagosomes or phagolysosomes containing electron-dense material that has the typical morphologic characteristics of nucleic acids [51].
Vacuolation appears to be greatest in the S1–S2 segments relative to the S3 segment and is consistent with the greater functionality of the vacuolar apparatus in the S1–S2 region, particularly in the S2 segment. The vacuoles likely a secondary phenomenon resulting from the extraction of oligonucleotide during processing and/or osmotic imbalances induced by the localized high concentrations of hydroscopic material in phagolysosomes. Oligonucleotides are highly water-soluble molecules and are extracted from tissue if the fixative used fails to completely crosslink the cellular components. In view of this, the high concentrations of oligonucleotides typically contained in cellular vacuolar apparatus would remain osmotically active during fixation, particularly with slower acting fixatives, such as formalin, as fixation in modified Karnovsky's solution or freezing does not lead to vacuole formation [58].
The cytoplasmic vacuolation observed in monkeys following treatment with 2′-MOE ASOs is consistent with an osmotic nephrosis. This type of renal change has been described for sugars, proteins, and pegylated proteins and also occurs during hypokalemia [59–61]. In each case, a nontoxic material (e.g., mannitol or sucrose) achieves a concentration in membrane-bound vesicles within the proximal tubular epithelium where influx of water leads to osmotic imbalances resulting in swelling of these solute containing vesicles, giving the histologic appearance of vacuoles. Ultrastructural examination differentiates this vacuolation with high concentrations of ASOs from degenerative vacuolation caused by nephrotoxicants, such as cisplatin, cyclosporine, mercuric chloride, cephaloridine, chloroform, and methotrexate [37,47,62–65].
In contrast to the swelling and vacuolation caused by high concentrations of ASOs, nephrotoxic compounds tend to cause widespread swelling of cytoplasmic organelles leading to the description of vacuolar degeneration as the osmotic balance of the entire cell is disrupted during the vacuolation process ultimately resulting in cell death. Generalized organelle swelling is not present in the tubular epithelial cells where an ASO has accumulated as granules or in vacuoles. Therefore, the vacuolation induced by ASOs is distinguishable from vacuolar degeneration. Furthermore, recent results indicate that oligonucleotide-containing vesicles develop into vacuoles during the handling and fixing of the tissues. As the oligonucleotides are concentrated in membrane-bound vesicles in vivo, there are osmotic differentials that lead to the development of this vacuolar artifact at the time of tissue harvesting and fixation. The best evidence for a role of tissue processing was that negligible vacuolation was seen when more rapid fixation with paraformaldehyde and glutaraldehyde was utilized even in animals given higher dose levels (40 mg/kg/week) [58]. As such, care needs to be taken in the handling and processing of kidney for histologic evaluation, and it is best to fix separate portions of the kidney cortex by immersion in both 10% formalin and 3% paraformaldehyde/3% glutaraldehyde (modified Karnovsky's solution).
Other dose- and concentration-dependent changes in proximal tubular morphology include minimal tubular epithelial atrophy, tubular epithelial regeneration, and epithelial cell degeneration at high doses with renal cortical concentrations of oligonucleotide >5,000 μg/g (Fig. 4). Minimal proximal tubular epithelial cell degeneration is a common dose-related finding frequently characterized by cytologic alterations or sloughing of individual tubular epithelial cells. These minimal changes would be expected with sequestering high concentrations of oligonucleotide in phagolysomes. At doses of 10 and 20 mg/kg/week 2′-MOE ASOs in monkey, the degree of cellular degeneration is minimal and not considered to be toxicologically important, based on the random distribution of single degenerative epithelial cells and the absence of associated functional alterations. At doses of ≥40 mg/kg/week, degenerative changes are of sufficient magnitude to change tubular functions most often detected by a slight increase in urine protein [57,58]. Importantly, vacuolation and any degenerative changes are reversible and are not associated with clinical pathology-based evidence of tubular dysfunction [58].

Relationship of ASO concentration in the renal cortex and type and severity of histologic alterations seen in monkeys (adapted from Henry et al. [1]). The bracketed area at concentrations <500 mg/g kidney cortex is the expected concentration range for clinical doses of 50–200 mg/week, which is far below the concentration needed to cause tubular degeneration in nonhuman primates. For reference, doses of 20 mg/kg/week in nonhuman primates lead to kidney cortex concentrations of 3,500–4,500 mg/g with all of the accompanying histological alterations.
Accumulation of drug in proximal tubules and the associated changes are not unique to 2′-MOE ASOs. PS ODNs at doses ≥40 mg/kg/week cause alterations in proximal tubular morphology at tissue concentrations ≥3,000 μg/g [57]. For 2′-MOE ASOs toxicologically relevant tubular degeneration occurs only at doses of ≥80 mg/kg/week and tissue concentrations ≥5,000 μg/g. The pathogenesis of the degenerative alterations at these very high doses is not known, but is most likely related to excessive accumulation substantive and high concentration of oligonucleotides in the proximal tubular epithelial cells (Fig. 4).
Histological alterations in the proximal tubular epithelium, including the presence of granules and cellular degenerative changes, are concentration dependent and the relationships between dose, concentration, and morphologic changes have been characterized [1]. Extrapolation of concentration–response relationships in monkeys to predict the relationship in humans is based on the similarities of the oligonucleotide pharmacokinetic profiles in monkey and man, and the pharmacokinetic profiles of 2′-MOE ASOs are generally predictable from one oligonucleotide to another [66].
Tubular morphologic changes and duration of treatment
Elimination of ASOs from the kidney follows first-order kinetics. As such, ASOs will continue to accumulate leading to increasing cellular concentrations until steady state is reached. Once steady state is reached, the degree of accumulation of ASOs in the proximal tubular epithelium does not increase in a meaningful manner with increasing duration of treatment in monkeys.
In the characterization of the toxicity profile, we observed that the duration of exposure/treatment was less critical in causation of toxicity than was cellular concentration. For example, the kidney cortex concentration is a more critical factor in determining the response of the renal tubular epithelium than the exposure time. As duration of treatment in monkeys with 2′-MOE ASOs increases from 1 to 3 months, renal cortical concentrations remained fairly constant and there was no worsening of morphologic changes in the kidney. As the changes in epithelial morphology are closely aligned with ASO concentrations and the proximal tubule epithelium has evolved to reabsorb and process solutes from lumen, it is logical that any effect would not be progressive. At more clinically relevant doses, the tubular changes were less frequent and less pronounced than that observed at the high dose. Furthermore, toxicity studies utilize dosing regimens with more frequent dose administration than those utilized in the clinical setting [58].
Assuming a renal half-life of ∼2 weeks, near complete clearance of ASOs from the kidneys takes about 13 weeks. Since these compounds follow first-order kinetics, 75% of clearance will occur after two half-lives, which is ∼1 month. As such, reversibility of histologic changes in the kidney occur once concentrations are <1,200–1,500 μg/g [66] (Fig. 4).
Clinical experience with PS ODN and 2′-MOE ASOs
The effects described above in preclinical models have not been observed in clinical trials with multiple sequences and a variety of pharmacologic targets. An integrated clinical/preclinical safety database developed by Isis Pharmaceuticals has facilitated detailed queries into different potential renal effects in >2,200 human subjects involved in multiple clinical studies. Overall findings in the renal safety database indicate no effects of PS or 2′-MOE on serum creatinine, blood urea nitrogen, or GFR, no changes in renal concentrating ability (specific gravity), no findings on microscopic examination of urine sediment, and no microalbuminuria in normal subjects or potentiation of preexisting microalbuminuria in type 2 diabetic subjects [67].
Influence of ASO Chemistry on Causation of Renal Effects
Other second-generation ASOs have further modified the backbone chemistry to influence stability and potency. The toxicity profile for many of these new chemistries varies slightly from the formulaic ASO “class toxicities” described for the 2′-MOE molecules, so sweeping statements on toxicity need to be framed by the particular backbone chemistry being considered. This is important as relatively minor modifications to the base pair sequence or backbone of an ASO can have dramatic effects on affinity and toxicity [68].
One such new chemistry is the LNA or locked nucleic acid. For mechanisms unknown, LNAs appear to be associated with more unexpected renal toxicities than other second-generation chemistries [69]. Acute tubular injury was reported in a healthy female volunteer in a first-in-human study with the LNA, SPC5001 [70]. Acute kidney injury (AKI) was evident shortly after the third once-weekly dose. Serum creatinine was increased and coincided with the appearance of white blood cells, granular casts, and hematuria on urinalysis. Renal biopsy indicated the presence multifocal tubular necrosis and ASOs. Following treatment for the AKI, serum creatinine levels gradually decreased in the patient and returned to baseline levels 44 days after the last dose of LNA. This individual fully recovered. Importantly, other volunteers who had received an equivalent dose of SPC5001 also had evidence of tubular dysfunction, providing additional evidence for a causal relationship for the AKI [70]. Notably, no untoward histologic or clinicopathologic effects were noted in the kidneys of cynomolgus monkeys given a loading dose of SPC5001 of 20 mg/kg followed by 4 weekly doses of 5 mg/kg.
In addition to tubular lesions, glomerulonephritis has been described in mice and monkeys with the 2′-OME PS ASO, drisapersen, in toxicity studies ≥3-month duration [71]. Renal alterations were present within 8 weeks following initiation of once-weekly subcutaneous dosing and were described as increased mesangial matrix and glomerular cellularity with occasional inflammatory cells or nuclear debris. An immune-mediated pathogenesis for the glomerular lesions in mice was put forward and supported by increased expression of inflammatory cytokines and other immunomodulatory genes in the renal cortex, increased production of CD68, and systemic increases of MCP-1. With treatment >6 months, however, immunoglobulin fragments mixed with amyloid was accumulated in the mesangium that was consistent with the murine syndromes of hyaline glomerulopathy and contributory amyloidosis. These alterations led to mortality and, in some mice, papillary necrosis secondary to vascular perturbation in the medulla. These chronic effacing renal lesions were interpreted to be caused by upregulation of murine-specific pathways and did not represent an alteration relevant to humans.
In contrast, examination of kidneys from monkeys following chronic drisapersen administration had microscopic glomerular lesions composed of increased mesangial and inflammatory cells and endothelial cell hypertrophy. Ultrastructurally, there were subepithelial and membranous electron-dense deposits that had characteristics of complement and complement-related fragments. The spectrum of lesions present in the monkeys most closely resembled C3 glomerulopathy, a condition that has been linked to dysregulation of the alternative complement pathway. In addition, anti-C3c immunofluorescence was noted along vascular tufts. Tubuloreticular inclusions were also present within the glomerular endothelium. This type of inclusion has been associated with immune deposition disease in humans. As such, inflammatory and/or immune mechanisms appear to be critical to the development of glomerular injury with species-specific sensitivities present in the mouse and monkey [71]. Notably, proteinuria with increased amounts of α1-microglobulin was seen in both a Phase 1 clinical study [72] and a Phase 2 clinical study with drisapersen [73] indicating that the animal findings were predictive of clinical effects on the urinary tract.
For perspective, none of the findings noted in the two examples above with LNA and 2′-OME ASOs has been seen in monkeys treated for 3 months with 12 different 2′-MOE ASOs. As the type of injury present in these cases was unrelated to what would be expected for pharmacodynamics target, lesions were most likely related solely to the chemistry of the ASOs. Furthermore, 9-month studies in monkeys have been conducted with five of these second-generation ASOs with no untoward effects on the kidney. Importantly, >2,000 patients and normal subjects have received doses of 2′-MOE ASOs with no demonstrable effects on renal function [67].
Summary and Conclusions
Single-stranded ASOs continue to mature as therapeutic agents through their ability to bind to mRNA and inhibit the production of toxic proteins. There is a spectrum of toxicities that has been observed commonly from sequence to sequence and although recent data suggest that there are sequence and modification differences, most ASOs fall with a predictable range of systemic effects in animals. In rodents and mice, the kidney contains the highest concentration of an ASO following parenteral administration and is the primary target organ for toxicity.
Some key features of the toxicity profile ASOs are as follows:
• Reabsorbed ASOs (basophilic granulation) may be accompanied by a cytoplasmic vacuolation artifact and is not regarded to be an adverse effect unless degenerative cytological alterations are present. • Cytoplasmic vacuolation in the proximal tubular epithelium is an artifact of fixation and can be prevented with perfusion fixation or immersion in more rapid fixative, such as paraformaldehyde/glutaraldehyde. • Production of degenerative alterations is concentration dependent and occurs at tissue concentrations ≥3,000 μg/g with PS ODNs and ≥5,000 μg/g with 2′-MOE ASOs, which is 10- to 100-fold greater than exposure at clinically relevant doses. • Once steady state is achieved, the degree of accumulation of 2′-MOE ASOs does not increase greatly with increasing duration of treatment. • Glomerular lesions are highly uncommon with PS ODNs and 2′-MOE ASOs and have only been seen secondary to activation of the alternate complement pathway cascading with activation of inflammatory cytokines and chemokines. • The toxicity profile for many of the new second-generation chemistries varies slightly from the formulaic ASO “class toxicities” described for the 2′-MOE molecules, so sweeping statements on toxicity need to be framed by the particular backbone chemistry being considered relatively minor modifications to the base pair sequence or backbone of an ASO can have dramatic effects on affinity and toxicity. • For mechanisms unknown, LNAs appear to be associated with unexpected AKI more than other second-generation chemistries. • The 2′-OME ASO (drisapersen) caused glomerular lesions in monkeys and mice secondary to complement and immune stimulation at doses much less than that necessary with 2′-MOE ASOs suggesting the 2′-OME ASO chemistry may be more prone to activate the alternate pathway of complement. Importantly, the interaction of 2′-MOE and the alternate pathway of complement has shown that nonhuman primates are much more sensitive than humans for activation of the pathway [74]. • Proteinuria with increased amounts of α1-microglobulin was seen in both a Phase 1 clinical study and a Phase 2 clinical study with drisapersen indicating that the animal findings were predictive of clinical effects on the urinary tract. Even though drisapersen is a fully modified backbone with a longer half-life, the 2′-OME ASO chemistry may be more prone to cause AKI than fully modified 2′-MOE ASOs.
As more second- and future-generation ASOs enter clinical development, more clinical–preclinical correlates may be identified. So far, findings in preclinical studies have not been predictive of effects seen in the clinical setting. The only exception to this statement is drisapersen, but renal effects were only fully appreciated in investigative studies following observations in clinical studies. Collectively, the data for PS ODN and 2′-MOE-modified ASOs have shown that the laboratory animal species utilized in toxicology studies generally overpredict renal effects of these agents. As such, 2′-MOEs do not appear to pose as much of a nephrotoxic risk to patients as the preclinical data would suggest. This observation has been confirmed so far in clinical investigations and continues to be monitored as more molecules enter clinical investigations.
Footnotes
Author Disclosure Statement
The author is an employee of Ionis Pharmaceuticals, Inc.