
Abstract
An active targeting drug delivery system that targets the nucleus could solve the problem of the treatment of genetic disorders through gene delivery, but it has met with limited success. The purpose of this study was to establish an RNA aptamer-modified nucleus-targeting liposomal carrier system referred to as NupApt-liposomes. RNA aptamers against the Nup358 protein are prepared using a newly established Protein SELEX method. After confirming aptamer binding to the recombinant protein, an aptamer–lipid conjugate (Apt-PEG-DSPE) was prepared. Aptamer-modified liposomes and simple polyethylene glycol (PEG) liposomes were prepared to check its ability to bind to isolated nuclei. Confocal studies indicated that the aptamer-modified liposomes had the ability to bind to isolated nuclei, whereas PEG-liposomes showed only weak binding. Confocal laser scanning microscopy studies of inhibition assays also supported the above conclusion. The dissociation constant of the Nucleoporin358-specific aptamer referred to as NupApt01 and NupApt02 were 36 and 70 nM, respectively. Finally, with aptamer-modified liposomes, gene expression studies showed a two times better gene expression in NupApt-liposome-treated nuclei in comparison to that of PEG-liposomes. This represents the first artificial RNA aptamer-modified liposomes promoting the specific binding of a nanocarrier to the nucleus, thus improving gene expression in comparison to PEG-liposomes.
Introduction
I
The nuclear envelope contains perforations, which are referred to as nuclear pores that participate in the import and export of macromolecules between the nucleus and cytoplasm. Each nuclear pore complex (NPC) contains eight filamentous nucleoporins called Nucleoporin358 or Nup358. NPC is a large complex of 125 MDa, acts as a barrier to macromolecules such as nucleic acids and various proteins, thus restricting their bidirectional traffic. The NPC opening is thought to be ∼39 nm in diameter and is composed of various copies of ∼30 different proteins called nucleoporins [1–4]. Nucleoporin358, also known as RanBP2, is the main and unique constituent of cytoplasmic filaments (∼50–100 nm long) of higher eukaryotic nucleus. The largest known nucleoporin is Nup358 and is composed of 3,324 residues (in humans). It interacts with karyopherins, other transport receptors, Ran, SUMOylated RanGAP1, and the NxF1-p15 mRNA export heterodimer, and hence mediates nuclear import and export. The first report of the exact location of RanBP2/Nup358 was published by Wu et al. [5]. Nucleoporin358 (Nup358) consists of several distinct regions, a ∼830 residue α-helical N Terminal Domain (NTD) followed by four Ran-binding sites, 8 Cys2-Cys2 type zinc finger motifs, an SUMO E3 ligase domain, phenyl-glycine (FG) repeats which are characteristic of nucleoporins and a C-terminal (CTD) cyclophilin A homology domain [5,6]. Apart from its essential role in nuclear transport, Nup358 also has other important functions. The upregulated expression of Nup358 was found in multiple myeloma tumorigenesis, an incurable malignant neoplasm, and mutation in the Nup358 gene is related to inflammatory myofibroblastic tumors and acute necrotizing encephalopathy [7–9]. HIV-1 capsid (CA) binds directly to the cyclophilin domain of Nup358/RanBP2, the C-terminus of Nup358/RanBP2 comprised of a cyclophilin homology domain mediates the docking of HIV-1 cores on NPC cytoplasmic filaments [10].
A nanoparticle-based drug delivery has been found to be a very effective nucleus drug delivery method in drug delivery system (DDS). Nanoparticle carriers must also overcome biological barriers similar to other drug delivery systems. However, attaching a ligand to a nanoparticle carrier can confer target selectivity to a nanoparticle. After escaping from endosome and its release into the cytoplasm, the pharmaceutical agent must deliver into the nucleus to achieve a therapeutic effect [10–15]. Many drug carriers composed of metals, oxides, semiconductors, and polymers can be used to achieve successful delivery, but the major concern is the access of the nanoparticles to the nucleus. The nucleus, a highly specialized controlling device of a cell, contains genetic material that is essential for cell survival. The main role of the nucleus is to contain and preserve hereditary material in an undamaged and intact state and to coordinate various cell activities with other cell organelles, such as cell division, growth, and protein synthesis. The nuclear envelope contains perforations, which are referred to as nuclear pores that participate in the import and export of macromolecules between the nucleus and cytoplasm. As the nucleus contains genetic material in a diseased state, it is essential for drugs to exert their action inside the nucleus to achieve a complete cure. Active targeting or receptor-based targeting of nanoparticles to different organelles in a cell has been successfully achieved in an attempt to permit therapeutic drugs to be taken up by cells and to reduce toxicity. After a review of recent progress in the field of nucleus drug delivery, it became clear that, a new nucleus-directed active targeting ligand would provide site-specific nucleus drug delivery and gene therapy. With this in mind, we attempted to target this most important organelle using an RNA aptamer ligand. Previously reported findings [16–25] suggest that it might be possible to develop a new ligand that can directly target the nucleus.
Aptamers are nucleic acids or peptide ligands that bind to various targets by adapting a 3D structure with a very high affinity and specificity. Unlike antibodies, they are nonimmunogenic, are smaller in size, and can be produced easily and cheaply. The SELEX method for selecting an aptamer from a random library pool by an iterative in vitro selection procedure was first reported by two laboratories independently in 1990 [26,27]. Viral and nonviral vectors both are used for successful gene delivery. The necessity of nonviral vectors increased after immunogenic issues rose on the use of viral vectors. The most exploited nonviral vector for nuclear drug delivery is a peptide sequence nucleus localization signal (NLS) [28]. Apart from NLS and its modifications, there was no nucleic acid-based ligand available. NLS is a natural ligand and has been extensively used for drug delivery, but the success ratio is limited. We, therefore, attempted to develop a new artificial nucleic acid-based ligand for this purpose. Increasing the success of an aptamer as a ligand prompted us to attempt to prepare a nucleic acid aptamer as artificial ligand to target the nucleus. We hypothesized that an RNA aptamer that binds to the nucleus could act as a ligand for artificial ligand-mediated active drug targeting (liposomal drug delivery).
Materials and Methods
General
Egg phosphatidylcholine (EPC), cholesterol (Chol), N-(lissamine rhodamine B sulfonyl)-1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (rhodamine-DOPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG-DSPE) were purchased from Avanti Polar Lipids. N-[(3-maleimide-1-oxopropyl) aminopropylpolyethyleneglycol-carbamy] distearoylphosphatidyl-ethanolamine (maleimide-PEG-DSPE) was purchased from Nippon Oil and Fat Co. Plasmid DNA encoding luciferase fusion protein (pCpG-free) and Octaarginine R8 was obtained from Sigma. Phosphatidic acid was purchased from Sigma.
Library preparation
A 99mer ssDNA synthetic template was initially purchased from Sigma-Genosys to prepare RNA. Eighty nucleotide ssRNA was in vitro transcribed with Durascribe T7 in vitro Transcription Kit (Epicentre Biotechnologies) (2 μL of water, 4 μL of 100 bp DNA template (1.0 μg), 2 μL of 10× reaction buffer, 2 μL each of 10 mM ATP, GTP, 2′-F-dCTP, 2′-F-dUTP, 2 μL 100 mM DTT, 2 μL of Durascribe T7 enzyme mix was incubated for 6 h at 37°C. One microliter (1 U) of DNase I was added and the suspension further incubated for 30 min. For RNA purification and precipitation, phenol: chloroform extraction and ethanol precipitation was performed. An equal volume of phenol-CHCl3-isoamyl alcohol (25:24:1) (100 μL), to in vitro transcription product was added and mixed well by vortexing, spun at room temperature for 2 min in the table top centrifuge. The aqueous layer was collected and an equal volume of chloroform was mixed well by vortexing and spun at room temperature for 2 min. The aqueous layer was separated, 0.1 volume of 3 M NaOAc, 1.0 μL glycogen (5 mg/mL), and a 2.5 volume of ethanol was added. This mixture was incubated at −20°C overnight to precipitate the RNA. The resulting solution was then centrifuged at 15,000 × g for 30 min at 4°C. The resulting RNA pellet was washed with 1 mL of 70% ethanol and spun again under the same conditions, the pellet was allowed to dry at room temperature, and was dissolved in DEPC-treated water. RNA was purified using an Illustra™ NAP-5 column chromatography (GE Healthcare) and concentrated with Millipore Amicon Ultra 0.5 mL filter units (MWCO; 3,000). The resulting RNA pool consisting of 20 nucleotide forward and reverse primer fixed regions at the 3′ and 5′ ends with 40 nucleotide central random sequence was used for selection.
Primer containing 5′ and 3′ template regions, T7 transcription promoter region is underlined-5′-AGCTCAATTC
Nup358-specific selection method
This method of selection is specifically designed to work well with all kinds of recombinant proteins. A 1.0 nmol library in the first selection and 500 pmol of RNA in subsequent selections was denatured in 1× selection buffer (50 mM Tris-HCl [pH 7.5], 5 mM KCl, 100 mM NaCl, 1 mM MgCl2, and 0.1% sodium azide). The 2′F-modified RNA library (1 nmol) was first heated in a thermo-block unit at 80°C for 10 min, and then cooled slowly to permit the secondary structures to form. The RNA library was then incubated with a nitrocellulose filter (0.45 μm; Millipore), the filter moistened with sterilized 1× selection buffer to remove filter-binding RNA species (referred as negative selection). The Nup358 protein contains 3,324 amino acid proteins and was provided as a generous gift from Dr. Shigehiro Yoshimura (Kyoto University). For positive selection, we used 824 amino acids of the glutathione S-transferase (GST)-tagged recombinant fusion Nup358 protein (specific sequence 2,501–3,324 amino acids, which is the C-terminal of human Nup358 or GST-Nup3582,501–3,324). After negative selection, the unbound RNA library was precipitated and incubated with recombinant Nup358 (62.5 pmol) in 1× SELEX buffer at 4°C for 30 min in an Eppendorf tube (500 μL). DISMIC mixed cellulose acetate ester syringe filter (0.20 μm; Advantech) was used for positive selection. After the incubation, the protein RNA mixture was loaded on the filter unit. Washing was done with 5 mL of wash buffer (2 mM HEPES-NaOH [pH 7.5], 3 mM MgCl2, 100 mM NaCl) and elution of protein-bound RNA using 3 mL of elution buffer (400 mM NaOAc, 5 mM EDTA, 7 M Urea) was performed. Upto seven rounds of selections, we performed two rounds of counter SELEX, to remove nonspecific binding, followed by positive selection. For the first counter selection at cycle no. 5, we used the same amounts of proteins (other than the target protein) such as bovine serum albumin (BSA), CA-1, and GST. In the seventh selection cycle, we introduced second counter SELEX and used cultured human cervical carcinoma cells (HeLa cells). The recovered library from the previous selection cycle was first subjected to negative selection and then was incubated with nontarget protein in the first counter SELEX and with cultured HeLa cells in the second counter SELEX at 4°C for 30 min. The unbound library was collected and incubated with the target protein to complete subsequent positive selection.
Amplification of the recovered library
Reverse transcription–polymerase chain reaction (RT-PCR) was performed with a QIAGEN One-Step RT-PCR Kit by following their procedure. 34.8 μL of water, 10 μL of (50 μM) 5× RT-PCR buffer, 2 μL of 10 mM dNTPs, 0.6 μL (50 μM) of reverse primer, 0.6 μL (50 μM) of forward primer, 1 μL of recovered RNA library, and 2.0 μL of RT-PCR enzyme mix were mixed and applied for PCR reaction. The sequence of forward primer was TCATAGGGAGGACAATCAGA and reverse primer was TGTACTCAGCGACGAGACTG. PCR conditions were: denaturation: 94°C, 30 s; primer annealing: 60°C, 30 s; and extension: 72°C, 1 min. A suitable cycle number was first investigated to prevent the amplification of undesired bands. Thirty-three nanomolar of 80 bp of DNA was recovered through phenol–chloroform extraction and ethanol precipitation and applied for the next PCR. Eighty base pairs dsDNA were amplified with PCR to prepare 100 bp dsDNA and the suitable cycle number was investigated. The sequence of the forward primer for second PCR was AGCTCAATTCTAATACGACTCATAGGGAGGACAATCAGA and the reverse primer was same as RT-PCR. Second PCR had 40.5 μL of water, 5 μL of 10× reaction buffer with MgCl2, 1 μL of 10 mM dNTP, 1 μL of (50 μM) reverse primer, 1 μL of (50 μM) forward primer, 1 μL of template cDNA, and 0.5 μL of Taq polymerase. PCR conditions: Denaturation: 94°C, 30 s; Primer annealing: 60°C, 30 s; Extension: 72°C, 1 min. A 3.8% agarose gel was prepared and a 1/10 fraction of PCR product was mixed with 1 μL of 6× loading buffer and loaded along with 20 bp ladder. The PCR-amplified DNA was resolved for 40 min at 100 V in 1× TBE buffer. The gel was observed under UV transilluminator (Kurabo). After staining in Ethidium bromide solution (1 μg/mL) for 15 min, the gel band with desired length of DNA was cut and chopped into small pieces. These pieces were mixed with 1× PBS and incubated on a shaking incubator overnight at 37°C. The extract was purified by Illustra™ NAP-5 column chromatography (GE Healthcare). The desalted DNA was purified again by phenol:chloroform extraction and precipitated by ethanol precipitation method. Conditions for colony PCR to identify transformed colonies were also fixed with finding suitable cycle number and amplified DNA was subjected to agarose gel electrophoresis.
Estimation of protein-bound aptamers and a gel shift assay
To estimate the recovered RNA after each selection, 1 μL of the selected libraries (filter binding and protein binding separately) were dissolved in 1× selection buffer and was subjected to NanoDrop machine (ND; Thermo Scientific) to get an accurate amount of RNA present in the solution.
For gel shift assay, the fluorescein amidite (FAM)-labeled RNA aptamer was heated at 80°C for 10 min and allowed to slowly cool to room temperature. A suitable amount of recombinant Nup358 protein and RNA aptamer were incubated at 37°C for 30 min. These mixtures were resolved in a 1.5% agarose gel in 1× TBE buffer at 100 V for 40 min. The gel was then exposed to the ImageQuant LAS4000 and band intensities were estimated with the ImageJ software. The equilibrium dissociation constant Kd was measured by fitting the dependence of the fluorescence intensity of unbound aptamer on increasing concentrations of the protein to the equation Y = BmaxX/(Kd + X) using the SigmaPlot 12 (Systat Software, Inc.).
Cloning and sequencing
The final and one of the most important steps of selection is the cloning and sequencing of the transformed colonies. To perform this experiment, the RNA library of the final selection was cloned (TOPO-TA Cloning Kit; Invitrogen) following the manufacturer's protocol. Briefly, the PCR product of the last selection was amplified with Taq Polymerase to add a single deoxyadenosine (A) to the 3′ ends. The PCR inserts were ligated in the vector followed by transformation. Luria Broth agar plates with antibiotics were prepared to grow transformed colonies. The transformed colonies were marked and identification was made by colony PCR. To determine the individual RNA aptamer sequence, DNA sequencing was performed. Homology among the entire sequences was analyzed using ClustalW software. The secondary structures of RNA sequences were determined using widely used Mfold software.
Synthesis of the RNA aptamer NupApt01 and NupApt02-conjugated Mal-PEG2000-DSPE
For the preparation of the aptamer-modified liposomes, we first prepared the aptamer–lipid conjugate. To prepare the conjugate, the individual aptamer NupApt01 and NupApt02 were mixed with nine times excess of Mal-PEG-DSPE in respective tubes. Briefly, 20 nM of aptamer (NupApt01 and NupApt02 separately) were initially incubated with 100 mM Tris-HCl and 200 mM DTT forming 200 μL reaction in water for 2 h at 37°C with continuous shaking on a Bio Shaker. Next, after 2 h, this solution was purified by means of an NAP-5 column (GE Healthcare). The purified product was incubated overnight with 180 nM of DSPE-PEG and 5% SDS at 37°C with continuous shaking on a shaking incubator. The next day, the conjugate was subjected to dialysis in a 3,000 MW cutoff dialysis membrane, submerged in 0.1% PBS, 0.5% SDS buffer overnight (the buffer was changed every 4 h for 16–20 h) to remove excess lipid. Next day, 50 mM sodium bicarbonate buffer was prepared and membrane was transferred to this buffer for the next 20 h (the buffer was changed every 4 h). The purified product was concentrated upto 100 μL with centrifugal concentrator (TOMY). The conjugation of aptamer to PEG-DSPE was quantified with spectrophotometer and confirmed by 3.8% agarose gel electrophoresis and quality was confirmed by high-performance liquid chromatography (HPLC; Elite Lachrom).
Preparation of liposomes
Liposomes are one of the most attractive carriers for successful drug delivery and are widely exploited in pharmaceutical research studies. All of the lipids were purchased from Avanti Polar Lipids. Liposomes (LPs), composed of egg yolk phosphatidylcholine (EPC): cholesterol (Chol) (7:3 molar ratio), was prepared by the lipid hydration method. In this method, all of the compounds were first added to a glass tube. A lipid film was allowed to form by evaporation of the solvents (chloroform and ethanol) in a vacuum until the solution completely evaporated. After ensuring that all the chloroform has dried up, the lipid film was hydrated with ice-cold HEPES buffer (10 mM, pH 7.4). The solution was incubated for 10 min at room temperature. The glass tube was then sonicated for ∼30 s in a bath-type sonicator (AU-25C; Aiwa). During the formation of the film, 1 mol% Rhodamine-DOPE was incorporated, to serve as a label for the lipid component. To modify the prepared liposomes with the aptamer–lipid conjugate, the required amount of preprepared lipid conjugate was added to the liposomes solution and incubated at 37°C for 1 h. To confirm if the modification of the aptamer conjugate over liposomes is successful or not, it was very important to check the size and zeta potential of modified and unmodified liposomes, which was confirmed by characterizing liposomes.
Characterization of liposomes
For experiments, several kinds of liposomes were prepared based on their ligand modification. To check the physical properties of liposomes, their size, PdI, and zeta potential values were measured. The mean size was measured using a quasi-elastic light scattering method, and the zeta potentials of the prepared liposomes were determined electrophoretically using Laser Doppler Velocimetry dynamic light scattering (Zetasizer Nano ZS; Malvern Instruments Ltd.). Liposome size and zeta potential of all types of liposomes were measured before and after conjugation.
Cell culture and nuclei isolation
For experiments, human cervical carcinoma cells (HeLa) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 μg/mL), and the cells were cultured under an atmosphere of 5% CO2 and 95% humidity at 37°C. One day before the experiment, 1 × 106 HeLa human cervix carcinoma cells (RIKEN Cell Bank) were cultured in DMEM. After removing the medium, the cells were washed twice with 1× PBS (0.1% BSA, 0.1% NaN3). One milliliter of 1× PBS (0.1% BSA, 0.1% NaN3) was added after washing and cells were scrapped with the help of a cell scrapper and collected in 1.5-mL Eppendorf tubes. All of the steps below were performed on ice. Isolation of washed cells was done by centrifugation (800 × g, 4°C, 2 min). After centrifugation, supernatant was removed and the pellet was suspended in ice cold HBSS 0.1% BSA. Then lysis buffer containing detergents NP-40 (HBSS 0.1% BSA, 0.2% NP-40) was added and cells were set on ice for 5 min. Again HBSS 0.1% BSA buffer was added in the solution and mixed gently by inverting the tube upside down two to four times followed by centrifugation at 800 × g, 4°C, 2 min. Supernatant was removed slowly without disturbing the pellet. These steps with lysis buffer were repeated until clean nuclei (white, delicate, less thick pellet) were obtained. As a final step, nuclei were resuspended in 1 mL of HBSS 0.1% BSA buffer. The quality of nuclei was checked under inverted microscope (Olympus U-CMAD3) and counting was done using hemocytometer with Trypan blue (0.3% v/v) staining method.
In vitro binding studies of aptamer-modified liposome to isolated nuclei
After the successful preparation of NupApt01 and NupApt02-modified liposomes and only polyethylene glycol (PEG) liposomes or nonmodified liposomes, nuclei were freshly isolated and used to check liposome binding. Twenty micromolar of aptamer-modified liposomes (NupApt01- and NupApt02-modified liposomes) were incubated in individual tubes containing ∼30,000 nuclei in a 200 μL reaction volume at 4°C for 30 min. After 30 min 1/10 of this mixture was separated in a 96-well-plate. Nuclei were centrifuged at 1,000 × g, 4°C, for 2 min, and the supernatant was removed. The precipitated nuclei were washed with 1× selection buffer and spun again under the same conditions. The nuclei were then resuspended in 1× selection buffer and 100 μL of the resulting mixture was added to a 96-well plate using Varioskan Flash 2.4 (Thermo Fisher Scientific).
Confocal laser scanning microscopy studies to study binding pattern of aptamer-modified liposome to isolated nuclei
Binding studies were performed by confocal microscopy (Nikon NIS Elements). At first, nuclei were isolated and counted. Separate tubes were prepared containing ∼30,000 nuclei in each. Twenty micromolar of NupApt01-modified PEG liposomes, NupApt02-modified PEG liposomes, and only PEG liposomes were added to the respective tubes. The nuclei–liposome mixture was incubated at 4°C for 30 min on ice and then spun on 1,000 × g, 4°C for 2 min. The supernatant was removed and nuclei were washed again using 1× selection buffer and spun. The nuclei were then stained by incubation with Hoechst33342 (1 mg/mL). Different glass slides were prepared and ∼300 nuclei were put over it and fixed with coverslip to observe binding pattern under confocal laser scanning microscopy (CLSM). The excitation/emission range of Rhodamine and Hoechst33342 were 530/586 nm and 350/461 nm, respectively. To check the specificity of liposome binding to nuclei, nucleolin (−) aptamer-modified liposomes were also prepared and their binding toward isolated nuclei was examined by CLSM using the same conditions in an individual experiment.
Inhibition assay
In this experiment, freshly isolated nuclei were first incubated with a 10-fold excess of aptamer-modified PEG-unlabeled liposomes for 30 min on ice, and the aptamer-modified PEG-labeled liposomes were then added and the resulting suspension was incubated for 30 min on ice. The ratio of unlabeled liposomes to labeled liposomes was (10:1). After incubation, the nuclei were washed and resuspended in HBSS 0.1% BSA buffer. Nuclei were incubated with Hoechst33342 (1 mg/mL) for 15 min and centrifuged at 1,000 × g, 25°C for 5 min to remove excess dye. Finally, the nuclei were fixed on a glass slide covered with coverslip and studied under confocal microscope (Nikon NIS Elements) at 100× magnification.
Kd value measurement of aptamer-modified liposome to isolated nuclei
Freshly isolated nuclei (∼30,000 per tube) were incubated with increasing concentration of ligand 0, 0.015, 0.031, 0.125, 0.5, 2.0, and 12.5 μM at 4°C for 30 min, after incubation nuclei were centrifuged to remove unbound liposomes in the supernatant. Nuclei were washed with HBSS 0.1% BSA buffer and centrifuged again. The supernatant was removed and pellet was resuspended in 0.1% HBSS 0.1% BSA buffer and put in 96-well plates serially as per ligand concentration. The binding affinity of NupApt01-modified PEG liposomes and NupApt02-modified PEG liposomes to nuclei was determined by measuring fluorescence intensity. The average mean fluorescence intensity of varying concentrations of aptamer-modified PEG liposomes was plotted to determine the dissociation constant Kd.
Assessment of transfection activity
HeLa cells (4 × 104) were seeded on a 24-well plate and cells were allowed to grow for 24 h. Three hours before transfection, media were changed with 250 μL of serum-free media (DMEM) and the cells were then set at 37°C. R8 MEND and aptamer-modified DOPE liposomes were added (9:1 ratio) to the cells and the resulting suspension further incubated for 1 h at 37°C. After the incubation, cells were washed once with 1 mL 1× PBS. One milliliter DMEM with 10% FBS was added to each well and incubated overnight in a CO2 incubator at 37°C. On the next day, the cells were washed with 1× PBS. Lysis buffer (Promega) was added followed by −80°C incubation for 30 min. Luciferase activity was initiated by adding 50 μL of luciferase assay reagent (Promega) into 20 μL of cell lysate followed by thaw at room temperature for few minutes. The luciferase activity was measured by a luminometer (Luminescencer-PSN; ATTO). The amount of protein was calculated by means of a BCA Protein Assay Kit (PIERCE).
Results
Enrichment of RNA aptamer library against Nup358 by protein SELEX
We established a new method referred to as the protein SELEX method, the steps of which were designed to specifically target recombinant proteins to produce nucleic acid aptamers. We used this SELEX method to generate RNA aptamers specific for a recombinant Nup358 protein (Fig. 1). The Nup358-specific protein SELEX included a sequence of negative and positive selections, and also at certain rounds, introduction of counter selections. With subsequent selections (after first selection) the amount of RNA library was decreased up to 1/10 from the initial amount to increase the strictness of the selection. The amounts of recovered RNA and filter-bound RNA were quantified by UV absorbance to check the enrichment after every selection round, and binding amounts were estimated (Table 1). For successful protein SELEX, it is mandatory to remove most of the filter-binding species and so it was important to monitor its constant loss after each selection. After five rounds of selection, a 9% library was found to bind recombinant Nup358, while filter-binding species could be removed successfully as the amount of filter-binding species after the fifth selection was under the detection limit. This indicates that the RNA, which could bind to the target protein, was amplified and enriched constantly. After the filter-binding RNA was removed successfully, the first counter SELEX was introduced to ensure the removal of any nonspecific binding sequences in the library. In the first counter selection the RNA library from the fifth cycle was first forced to a negative selection and the unbound RNA library was then recovered and incubated with CA I, GST, and BSA protein. After incubation, the recovered unbound RNA was then used for subsequent regular selection. In seventh (final round) selection, a second counter selection was introduced using cultured HeLa cells. For this experiment, after the seventh negative selection the generated aptamer library was incubated with cultured HeLa cells and the unbound library was recovered and precipitated. On completion of the seventh selection, as per UV absorbance data ∼11% of the RNA library was recovered as recombinant Nup358-bound RNA.

Schematic diagram of protein SELEX for Nup358 protein. The RNA aptamer library was subjected to negative selection followed by subsequent positive selections. In the fifth and seventh SELEX, counter selection was introduced using nontarget proteins and cultured HeLa cells, respectively. The nonbinding aptamers were collected and incubated with the target Nup358 protein for positive selections. After elution, the target-binding RNA aptamers were amplified by PCR. After successful enrichment of the aptamer library, DNA sequencing was performed to identify individual sequences. PCR, polymerase chain reaction.
Identification of the strongest aptamer candidates
The major step was to identify dominantly enriched RNA aptamer candidates, once the selection cycles were completed. For this, RNA library after the seventh selection and successful enrichment, Nup358 was cloned using the TOPO-TA vector. The transformed colonies were identified by colony PCR and sequenced. Homology among the whole sequences was analyzed using the ClustalW software and a guide tree was prepared (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat). Out of 50 readable sequences, seven distinct sequences were identified in multiple copies (Table 2). To understand the structure of the aptamers, secondary structures were predicted using the Mfold software (Supplementary Fig. S2). It was essential to confirm the binding of these aptamer sequences to the target protein and so to confirm the binding of all seven obtained distinct sequences, as a first step, dsDNA from the respective pDNA was prepared and purified. The dsDNA was in vitro transcribed and obtained RNAs were applied for electrophoretic mobility shift assay (EMSA) (Fig. 2A). From that result, two of them (NupApt01 and NupApt02) showed a better binding ability than the others, as a smaller amount of protein produced better shift and lesser free aptamer on the gel. On further investigation, we found that the RNA sequence possessed no affinity for other proteins such as BSA; also no affinity of 40-nucleotide FAM-labeled random library for recombinant Nup358 protein was observed (Fig. 2B, C). The specific complex formation of the recombinant Nup358 protein and the NupApt02 RNA aptamer confirmed that a single-stranded RNA aptamer specific for the recombinant Nup358 protein was produced successfully. The dissociation constant (Kd) of NupApt02 was calculated as 1.6 μM using a gel shift-binding assay (Fig. 3).
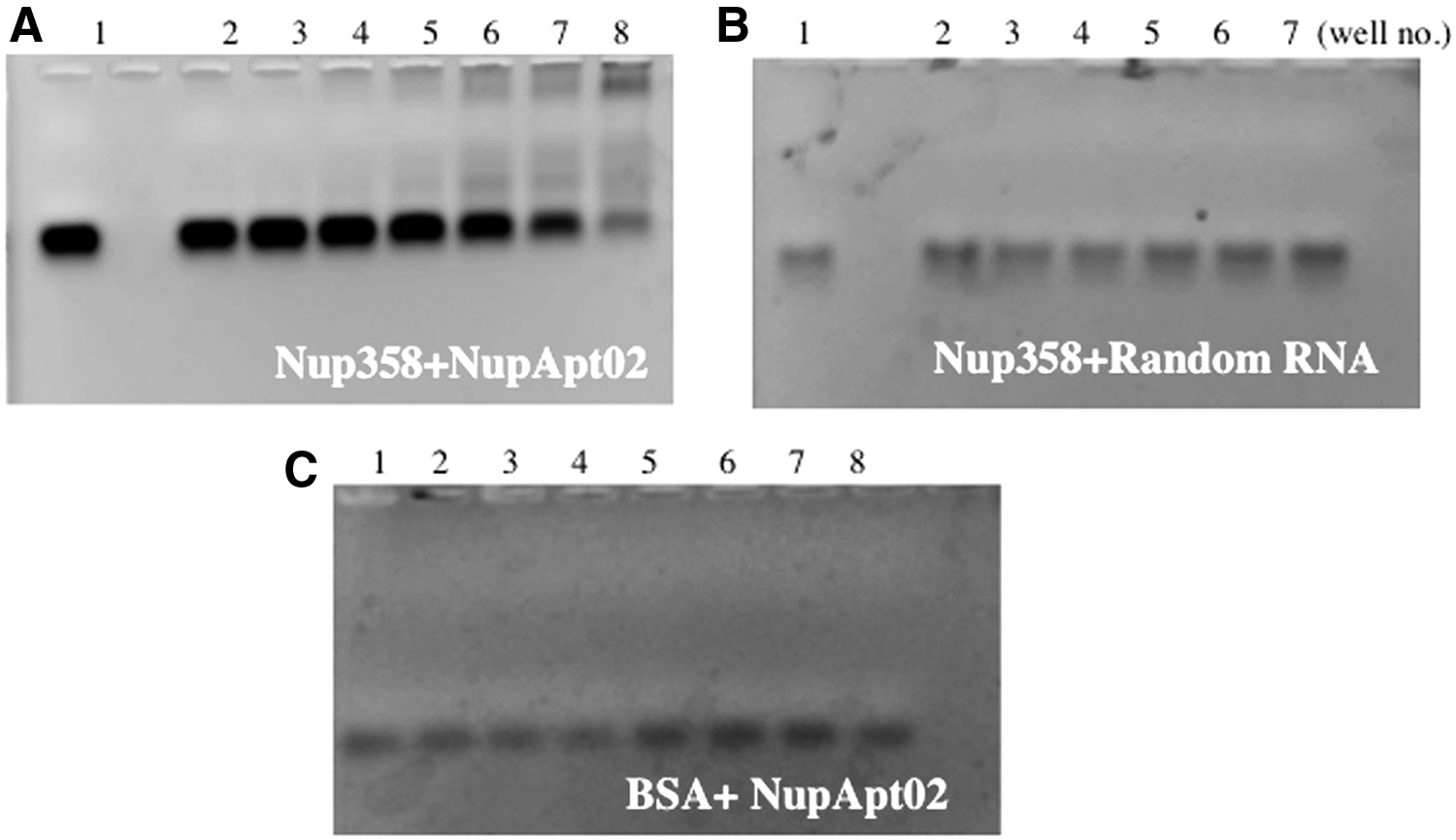
Selective binding of the aptamer NupApt02 to Nup358 protein. EMSA was investigated with

Determination of the binding affinity (Kd value) of the RNA aptamer NupApt02. FAM-labeled aptamer and RNA aptamer NupApt02 (50 nM) with increasing concentrations of target protein in 1× selection buffer was incubated on a scale of 35 μL. Gel shift assay was performed on a 1.5% agarose gel, 30 min at 100 V. The gel analysis was done under ImageQuant LAS 4000 and the binding was measured by quantifying and calculating the value of free RNA aptamer using ImageJ software. Kd value was 1.6 μM. BSA protein was used as negative control.
Sequences were multiple aligned using ClustalW software. Numbers denote copies of individual sequence in total number of sequences.
Preparation of aptamer-modified liposomes with postinsertion method
For the establishment of an aptamer targeted liposomal drug delivery system, it was necessary to prepare aptamer-modified liposomes. To prepare aptamer-modified liposomes, a conjugate was first prepared and the conjugated Nup358-specific RNA aptamer NupApt01 and NupApt02 with maleimide DSPE-PEG were then modified over the prepared PEG liposomes. Farokhzad et al. in 2004 first introduced such an approach [29]. The conjugation of the NupApt01 or the NupApt02 aptamer with Mal-PEG-DSPE was done in 10 mM Tris-HCl buffer and was prepared successfully. A 3.8% agarose gel confirmed the conjugation by electrophoresis followed by gel staining. The conjugate, being of higher molecular weight, could not move faster on the gel (Fig. 4). After preparation of the conjugate it was essential to modify it over liposomes. Next, EPC:Chol (7:3 molar ratio) liposomes were prepared by the lipid hydration method. Adding the compounds to a glass tube and allowing the mixture to dry under a vacuum formed the lipid film. The completely dried film was hydrated with buffer followed by incubation and sonication. The required amount of aptamer–lipid conjugate was mixed with the liposomal solution and incubated. To confirm that the liposomes were successfully modified with NupApt01- or NupApt02-modified PEG-DSPE conjugate, the average size in diameter and zeta potential of the prepared liposomes was measured (Table 3).
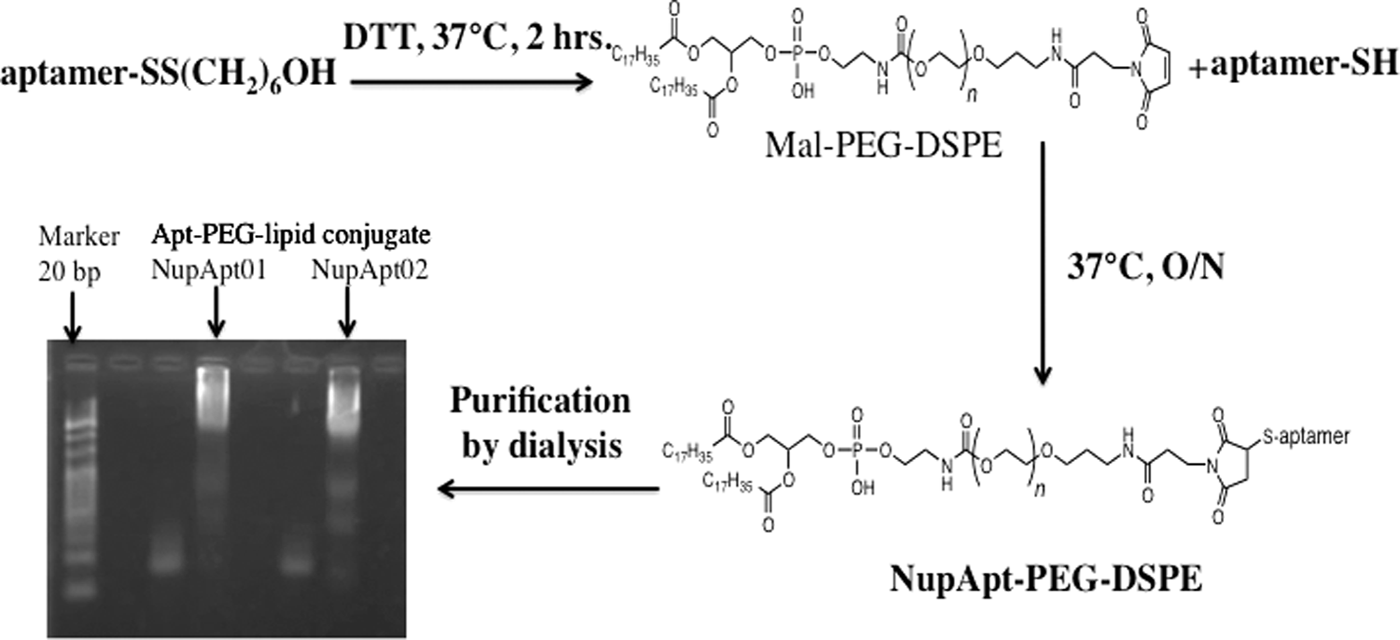
A schematic diagram of the conjugation reaction of NupApt-PEG-DSPE-lipid. Lane 1 is 20 bp DNA marker, 3 is NupApt01-lipid before conjugation, 4 is Apt-PEG-lipid conjugate, 6 is NupApt02-lipid before conjugation, 7 is Apt-PEG-lipid conjugate, and 2, 5, and 8 are empty lanes. PEG, polyethylene glycol.
PEG, polyethylene glycol.
Evaluation of the binding of aptamer-modified liposomes to nuclei
To confirm that the aptamer-modified liposomes could bind to nuclei in vitro, liposomes were treated with nuclei isolated from HeLa cells. Fixed numbers of nuclei were incubated with aptamer-modified and nonmodified liposomes for 30 min on ice. Nuclei were washed with selection buffer and a small amount of this solution was used for fluorescence intensity measurements. At the end of the experiment, it was found that both the aptamer-modified liposomes, NupApt01-modified PEG liposomes and NupApt02-modified PEG liposomes, showed significantly higher nucleus binding in terms of fluorescence intensities, whereas the nonmodified or PEG-liposomes failed to show strong binding (Fig. 5). The NupApt01-PEG-liposomes and NupApt02-PEG-liposomes showed seven times and six times better binding to the nucleus in comparison to nonmodified liposomes.
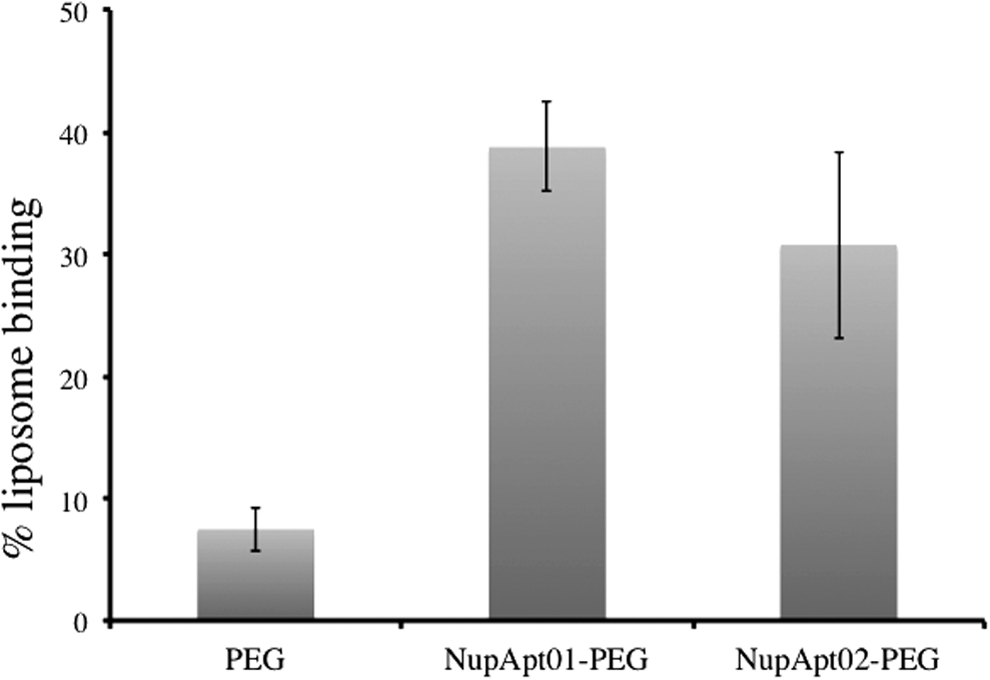
Aptamer-modified liposome-binding studies using isolated nuclei. FI results showed seven to six times better binding of NupApt01 (20 μM) and NupApt02-modified liposomes (20 μM) to isolated nuclei (30,000 in numbers), in comparison to PEG liposomes (20 μM). FI, fluorescence intensity.
Imaging of the binding of aptamer-modified liposomes to nuclei and estimation of the dissociation constant of aptamer-modified liposomes
Liposomes are carrier systems that take drugs to a specific intracellular site. However, despite several gene/drug delivery attempts, the direct, aptamer-mediated liposomal targeting to a nucleus has not been reported. After confirming that the aptamer-modified liposomes show better binding compared with nonmodified liposomes in vitro, as evidenced by fluorescence intensity results (in comparison to nonmodified liposomes), it then became important to perform experiments to produce visible results that confirmed the localization of NupApt01, NupApt02, a negative control sequence, and PEG-modified liposomes in nuclei, isolated from the HeLa Cells. For a negative control aptamer, we employed a nucleolin-negative aptamer (CCTCCTCCTCCTTCTCCTCCTCCTCC), which was originally reported by Kotula et al. [30]. This aptamer sequence does not bind to the nucleus and liposomes modified with this negative aptamer sequence should also not bind to the nucleus surface. Liposomes were prepared by the hydration method as described earlier and stained with Rhodamine and their physical properties were measured by a Zetasizer (Table 3). After characterization, these liposomes were incubated with isolated HeLa nuclei, which were stained with Hoechst33342. The binding characteristics of these liposomes were analyzed by CLSM. As a result, CLSM studies clearly showed the specific binding of the NupApt01-modified liposomes and NupApt02-modified liposomes to the nucleus, compared with nonmodified liposomes (Figs. 6 and 7). NupApt01- and the NupApt02-modified liposomes were found to have accumulated around the nucleus, whereas, as expected, the negative control and PEG liposomes failed to bind to the nucleus. The target protein Nup358 is cytoplasmic, an exposed fiber-like extension (a few nano meters in length, eight in number per NPC) attached to the nuclear membrane through the N-terminal domain and so, based on the CLSM results, we concluded that Nup358-specific RNA aptamer-mediated liposomes are able to bind specifically to the nucleus because the aptamer-modified liposomes were found to have accumulated around the rim of the nucleus, which is the site of nucleoporins358, whereas other sequences or PEG liposomes failed to bind specifically or strongly.

CLSM studies of the binding of NupApt-PEG-LPs bound to the isolated nucleus. NupApt01-PEG-LPs (20 μM) and NupApt02-PEG-LPs (20 μM), nucleolin (−) PEG-LPs (20 μM) and PEG-LPs (20 μM) showed different binding patterns according to their specificity. Nuclei were 30,000/tube in number and were stained with Hoechst33342. Scale bar = 10 μm. CLSM, confocal laser scanning microscopy.
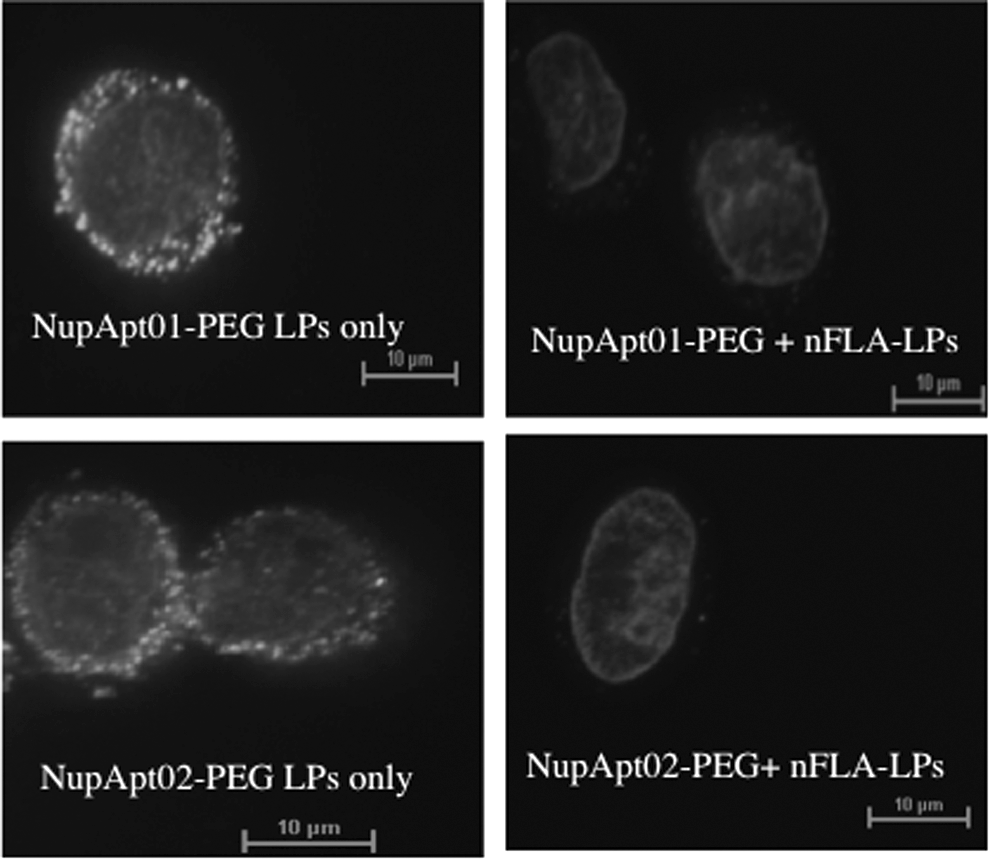
Inhibition assay. NupApt01-PEG-LPs and NupApt02-PEG-LPs (labeled) in the absence of excess of nFLA-LPs or inhibitory liposomes showed strong signals, whereas pretreatment of isolated nuclei with labeled liposomes reduced labeled liposomes binding drastically. Nuclei were 30,000/tube in numbers and were stained with Hoechst33342. Scale bar = 10 μm. nFLA-LP, nonfluorescent-labeled aptamer.
The estimation of the dissociation constant of liposomes would constitute a good parameter for discussing the efficiency of nucleus targeting. The dissociation constant of the aptamer-modified liposomes to nuclei was calculated based on fluorescence intensity measurements. Different concentrations of aptamer-modified liposomes were incubated with fixed numbers of isolated nuclei, the liposomes were allowed to bind to the nuclei, and the suspension was then centrifuged to remove unbound liposomes. The nuclei pellet was washed and then resuspended. Fluorescence intensity was measured using a fluorophotometer. A dissociation curve was plotted by measuring the average mean fluorescence intensity of varying concentrations of aptamer-modified PEG liposomes. The dissociation constant of NupApt01-modified liposomes was calculated as 36 nM, and NupApt02-modified liposomes were 70 nM (Fig. 8), respectively, which showed a higher dissociation constant compared with a single aptamer.

Dissociation constant of NupApt-modified PEG liposomes. Isolated nuclei (∼30,000) were incubated with increasing concentration of ligands (0–12.5 μM), for 30 min, 4°C. Fluorescence intensity measurement data to check dissociation constant of
Investigation of in vitro binding of aptamer-modified labeled liposomes in the presence of nonlabeled liposomes or inhibition assay
If aptamer-targeting liposomes bind to the nucleus in a ligand–receptor-mediated manner, it must show saturation after all receptors were close to being occupied. To test this hypothesis, a saturation binding assay was performed. Aptamer-modified nonfluorescent-labeled and aptamer-modified fluorescent-labeled liposomes were prepared. Freshly isolated nuclei from HeLa cells were first incubated with a 10 times excess of nonfluorescent-labeled liposomes after incubation, fluorescent-labeled liposomes were added, and the suspension further incubated. CLSM results confirmed that on the preincubation of nuclei with aptamer-modified nonfluorescent-labeled liposomes, no fluorescence was detected on the nucleus surface, indicating that the aptamer-modified liposomes saturated the specific aptamer-binding sites of the nucleus. As a result, the aptamer-modified fluorescent-labeled liposomes could not bind further. This confirms that the binding of Nup358-specific aptamer-modified liposomes on nuclei was ligand mediated (Fig. 7).
Transgene expression of aptamer-modified MEND
To investigate the effect of aptamer-targeted liposomal drug delivery, luciferase-encoded pDNA was encapsulated and delivered to HeLa cells. The pDNA was encapsulated in DOPE/PA (7:3 molar ratio) liposomes and then modified with the NupApt01, NupApt02, or PEG. Conventional 10% R8 liposomes [31] were prepared and an aptamer-modified MEND and R8 liposomes were mixed together in 9:1 ratio (R8 liposome: aptamer liposome = 9:1 molar ratio) and transfected for 1 h. Gene expression was two times higher in case of NupApt01 MEND and 1.5 times higher in NupApt02 MEND in comparison to nonmodified MEND (Fig. 9). The overall scheme to explain entry of aptamer-modified liposomes into cell is described (Fig. 10).

Luminometer's luciferase activity data on uptake efficacy of each liposomal system. Two individual experiments with three copies of each sample, the mean value was used in calculating luciferase activity, and gene expression with standard deviation being used in calculation. The amount of protein was estimated with BCA protein assay.

Schematic presentation of aptamer-mediated nucleus targeting the drug delivery method.
Discussion
In this study, we report on the preparation of an RNA aptamer that binds to the Nup358 protein in vitro. We successfully established a new selection method, referred to as protein SELEX, for the selection of all kinds of recombinant proteins in vitro. The selection was first carried out with another protein CA I as we reported earlier [32], and then with the Nup358 recombinant protein, and we found that this method can be used for any specific protein to generate aptamers. We started the selection with a complex 2′F base modified random RNA library containing 20 nucleotide fixed regions at the 3′ and 5′ positions and a 40 nucleotide random region in between. Removal of filter-binding species was required for successful selection and so it was very important to perform a negative selection before each selection. To remove nonspecific binding materials it was important to introduce a counter selection after a few selections. Most importantly, we were able to improve the cloning and sequencing results by introducing negative and counter SELEX. After the fifth cycle of selection, we performed sequencing and found two conserved sequences, whereas the amount of RNA library binding to Nup358 protein was 9.8%. This means that there was scope for removing RNAs that bind to Nup358 protein with electrostatic, hydrogen bonding or hydrophobic interactions. To get better aptamers, it was necessary to remove nonspecific sequences. To accomplish this, we carried out two more selections and introduced a second counter SELEX with cultured HeLa cells. After we completed the second counter selection and the seventh selection, the amount of bound RNA was reduced to 11% compared with the starting RNAs.
To confirm the library enrichment, we applied this enriched library to sequencing and in 50 sequences seven different kinds of conserved sequences were obtained in multiple copies (38% of total sequences were conserved). NupApt02 was a sequence that repeated after the fifth selection and after the seventh selection cloning result. Under rigid selection conditions, the NupApt02 sequence survived till the end, meaning that it is a strong aptamer candidate. NupApt01 was one of the conserved sequences, which was identified in six copies making it an even stronger candidate. During the selection, it was difficult to add any other protein to remove nonspecific binding species because it could result in the generation of an RNA library that has binding affinity for other proteins (nontarget) added during the selection. In the case of the addition of several proteins during selection, the RNA library itself cannot identify between a target and nontarget protein. Because of this, we did not add any other protein during the selection, and so it was necessary to perform a counter selection after a few rounds of selection. Counter selection as per our experience should be performed after at least two or three rounds of selection, because the complexity of the library may decrease drastically if counter selection is done in very initial cycles.
We also found that it was essential to monitor the increasing or decreasing percentage of filter-binding RNA library. It must decrease constantly after the initial selection and be completely removed after a few selections. In initial cycles, if it is not possible to be removed efficiently, it may result in the failure of selection. After seven selection cycles we successfully removed nonspecific binding compounds as well, such as filter-binding species, and were able to generate two strongest RNA aptamers, NupApt01 and NupApt02, specific for recombinant Nup358. EMSA was employed to verify the RNA aptamer library binding and after sequencing, the selected individual aptamer binding to the Nup358 protein. Six out of seven sequences were found to bind to protein. It was found that, on agarose gel electrophoresis, the protein–RNA complex failed to migrate at all and the band always appeared close to the agarose gel well. This was confirmed by locating the position of the protein in agarose gel by Coomassie Brilliant Blue (CBB) staining. Free RNA in the gel is also a significant parameter that can be used to confirm the percentage binding of an RNA library to a protein. The same concentration of BSA protein was used to check the nonspecific binding of the NupApt02 aptamer, but no shift was observed on the gel. In addition, when a random FAM-40 nucleotide RNA library was applied to an agarose gel with the CA I protein, no shift was detected. This confirms the specificity of the NupApt02 RNA aptamer for the Nup358 protein. We estimated the dissociation constant from EMSA for the FAM-tagged aptamer and the Nup358 protein and the Kd value was determined to be 1.6 μM. Our protein SELEX method is an economical, convenient selection procedure that can produce high-affinity aptamers for two different proteins (in our case Nup358 as well as CA I). Since there are naturally occurring and modified ligands, to our knowledge, this nucleic acid aptamer that binds specifically to a nucleus-related protein and can be used to enhance nuclear import, is the first attempt in the area of artificial ligand-mediated nuclear drug delivery.
In this study, we hypothesized that it would be possible to prepare recombinant Nup358 protein-specific RNA aptamers that can identify a specific protein on the nuclear membrane. The newly established protein SELEX method was used and a series of RNA aptamers were generated and identified, which were found to be acceptable for use as a ligand for active nuclear-targeted drug delivery. The aptamer–lipid conjugate was first prepared and liposomes were then prepared separately. The liposomes were then modified with an aptamer–lipid conjugate, and, like this aptamer, modified PEG liposomes were prepared by the postinsertion method. The two nucleus-specific RNA aptamers, NupApt01 and NupApt02-modified PEG-liposomes, showed very strong binding (six to seven times) toward the nucleus, whereas PEG-liposomes were not bound strongly to the nucleus. Confocal microscopy studies confirmed the binding of the aptamer-modified liposomes to the nuclear periphery. Whether the binding of aptamer-modified liposomes was specific or not was confirmed by comparing the binding of nucleolin (−) apt-PEG-liposomes and aptamer-modified PEG-liposomes. The results of CLSM images clearly showed that nucleolin (−) apt-modified liposomes did not bind to the nucleus. An inhibition assay confirmed that the binding of liposomes to nuclei is ligand mediated, as on the addition and preincubation of nuclei in the presence of an excess of aptamer-modified unlabeled liposomes, the binding of the later added aptamer-modified labeled liposomes was drastically reduced. The Kd value of a single aptamer was not very strong, but aptamer-modified liposomes had a very good binding affinity. An RNA aptamer binds to its target protein through a size fitting mechanism; so only a specific aptamer or an aptamer-targeted drug carrier should bind to its specific site. The aptamer forms a decoration over the liposome outer surface, and as a result, the aptamer first identifies the target region, forms a complex, and then becomes attached to the surface of the nucleus.
Aptamer-modified liposomes were first prepared, and in comparison to nonmodified liposomes and negative aptamer-modified liposomes, showed strong binding signals toward isolated nuclei and, as a result, showed a strong Kd value on the basis of aptamer liposome binding to the nucleus, and a decrease in the dissociation constant of a single aptamer and aptamer-modified liposomes from 1,600 to 36 nM was observed. Based on a report by Wilner et al. [33], the binding of a transferrin-specific RNA aptamer was improved by 300 times (104 nM to 310 pM) when the aptamer was modified through a liposome, as the aptamer present on the liposome surface provides multivalent binding toward its target [33]. Similarly in a different report, Kibria et al. reported that large-sized liposomes modified with a specific ligand provide multivalent binding [34]. Because, in a series of steps, we prepared EPC:Chol liposomes and later an aptamer–PEG conjugate was modified through postinsertion, it is thought that several specific ligands (aptamers) present over the nanoparticle permitted the nanoparticle to target a protein more specifically and strongly with better binding probability due to the existence of multivalent binding.
Since the target of all drug delivery systems is to ultimately reach the nucleus, either directly or indirectly, this system constitutes a new development in the field of nuclear-targeted DDS. There have been several reports on NLS-based nucleus targeting, but the number of successful targeting are very low. The idea of designing an artificial ligand that can mimic the role of NLS and possibly in future can provide better targeting was a need. Our report as per our knowledge represents the first successful attempt in the area of artificial ligand-mediated nuclear-targeted drug delivery directly to the nucleus. It is a common understanding that in an actively dividing cell the transfected plasmid/cargo reaches the cytoplasm and enters the nucleus following mitosis, when there is a nuclear membrane breakdown and formation. Our current finding adds value in cancer research, as this approach may be more useful in nondividing cells, where the nuclear membrane breakdown does not occur and efficiency of nuclear uptake is low. The aptamer-modified liposomes are well attached to the nucleus membrane and release the drug into the nucleus through membrane fusion. Thus, this technique is a ray of hope in cancer research and treatment. However, further studies will be required to acquire sound detailed knowledge regarding aptamer-modified liposomes that can function efficiently in systems, such as through in vivo studies. As a result, it can be concluded that a new SELEX method was successfully developed, along with first RNA aptamers that specifically bind to the nuclear Nup358 protein. These aptamers are pioneer artificial ligands for nuclear-targeted delivery that shows liposomal binding in intracellular experiments using isolated nuclei.
Nanoparticle–aptamer conjugation is a new approach to facilitate the delivery of nanoparticles to a specific site of action. Our finding is a continuation of this work that facilitates the binding of a nanoparticle–aptamer bioconjugate directly to the nucleus. However, aptamer-based nanoparticles must be appropriately engineered to work perfectly after systemic administration to avoid toxicity. Such modifications include controlling optimal size of nanoparticles and surface modifications. The findings reported herein clearly show that our RNA aptamer-8-modified vehicles bind efficiently to the nuclear surface protein Nup358 and that a nanocarrier directed by this aptamer can also be used for direct nuclear targeting and drug delivery.
Conclusions
This is the first demonstration to show that liposomes modified with aptamers (ligand) through a short PEG stretch can specifically bind to the surface of nuclei derived from HeLa cells. The experimental results suggest that a high-affinity ligand could be prepared that has an affinity for a specific nuclear Nup358 protein that is present at the cytoplasmic phase of the nucleus, in a flagella-like structure. To accomplish this, a new selection method was established referred to as Protein SELEX for producing high-affinity RNA aptamers for recombinant proteins in vitro. After seven rounds of selections, cloning and sequencing was performed, the sequences were aligned, and seven sequences were identified. EMSA results suggested that two of seven sequences showed very strong binding to the recombinant protein and no nonspecific binding could be observed. After the identification of aptamers, they were next used as a ligand to modify liposomes by a postinsertion method. Fluorescence intensity results confirmed that aptamer-modified liposomes have six to seven times better binding capability toward isolated nuclei in comparison to PEG liposomes. CLSM studies confirmed that the aptamer-modified liposomes bound specifically to the nuclear membrane, whereas PEG liposomes did not show binding. Nucleolin (−) aptamer-modified liposomes did not show binding when incubated at a high concentration, indicating that the binding was specific. An inhibition assay was performed to confirm that the binding of liposomes to nuclei is ligand assisted, and on adding an excess of aptamer-modified nonlabeled liposomes, the binding of aptamer-modified labeled liposomes decreased. The dissociation constant of aptamer-modified liposomes were 36 and 70 nM (Apt01 and Apt02, respectively), both of which bound well on nucleus, but based on all of our experimental results and finally the Kd value results, the NupApt01 appears to be the stronger binding aptamer.
Taken together, the findings provide a proof of concept for nuclear drug delivery. An RNA aptamer was used for the first time as a ligand, to target Nup358 (a surface-specific protein of the nucleus) for assisting the binding of nanoparticles to the nucleus. Therefore, this new work may offer a new strategy for the discovery and development of nuclear drug delivery systems. As gene therapy is the ultimate goal of therapeutics, this work may fill the gap between assumptions and facts.
Footnotes
Acknowledgments
Special Education and Research Grant of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan supported this work. The authors thank Dr. M.S. Feather for his expert advice in writing the English article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.