
Abstract
Lentivirus vectors are presently the favorite vehicles for therapeutic gene transfer in hematopoietic cells. Nonetheless, these vectors integrate randomly throughout the genome, exhibiting variegation of transgene expression due to the spreading of heterochromatin into the vector sequences. Moreover, the cis-regulatory elements harbored by the vector could disturb the proper transcription of resident genes neighboring the integration site. The incorporation of chromatin insulators in flanking position to the transferred unit can alleviate both the above-mentioned dangerous effects, due to the insulator-specific barrier and enhancer-blocking activities. In this study, we report the valuable properties of the sea urchin-derived sns5 insulator in improving the expression efficiency of a lentivirus vector integrated in the mammalian erythroid genome. We show that these results neither reflect an intrinsic sns5 enhancer activity nor rely on the recruitment of the erythroid-specific GATA-1 factor to sns5. Furthermore, by using the Chromosome Conformation Capture technology, we report that a single copy of the sns5-insulated vector is specifically organized into an independent chromatin loop at the provirus locus. Our results not only provide new clues concerning the molecular mechanism of sns5 function in the erythroid genome but also reassure the use of sns5 to improve the performance of gene therapy vectors.
Introduction
I
Although maintaining expression of the integrated transgenes is a prerequisite for the clinical success of gene therapy, the use of transgenes potentially conceals dangerous genotoxicity drawbacks. In this respect, the cis-regulatory sequences harbored by the vector can interfere with the proper transcription of resident genes surrounding the integration site. This occurrence leads, in the most extreme cases, to malignant transformation of a given cell clone, as reported in the first clinical trials employing γ-retroviral vectors (reviewed in Refs. 13–15).
Lentiviral vectors (LV) represent a more promising tool for human gene therapy, owing to their ability to transduce primary nondividing cells [16–19] and the overall reduced mutagenic aptitude compared to their γ-retrovirus counterparts [20,21]. Nonetheless, lentivectors also display enhancer-mediated dysregulation of neighboring genes in animal models of metabolic diseases [22,23]. Consequently, a strong effort is currently directed toward improving the safety of therapeutic LV.
A growing body of evidence shows that the incorporation in the transferred unit of a class of specialized cis-regulatory elements known as chromatin insulators can reduce vector–genome reciprocal interactions, mitigating both vector silencing and vector-mediated genotoxicity [13,24–27].
Identified in a wide range of metazoans, chromatin insulators exhibit two key properties. They can block enhancer–promoter communication only when interposed between them and can act as barrier against the spread of repressive chromatin (28–30, recently reviewed in Refs. 31,32). The sequences that mediate these activities appear to be mechanistically distinct and physically separable [10].
In addition to these functions, insulators are thought to play a key role in shaping the three-dimensional genome organization. Indeed, high-resolution mapping of chromosomal domains suggests their involvement in the establishment of local chromosome environments as well as in long-range chromosomal interactions in Drosophila and higher eukaryotes [33–37].
A DNA element displaying insulator features, sns5, has been identified at the 3′ end of the sea urchin Paracentrotus lividus H2A gene, within the tandem repeat of the early histone unit [38]. We demonstrated that sns5, when tested in the natural genomic context, shields the downstream H1 promoter from the activity of H2A enhancer, suggesting that sns5 is involved in the differential accumulation of nucleosomal and linker transcripts during embryogenesis of the sea urchin [39,40]. Strikingly, the sns5 insulator activity is conserved in various cell types and across species [41–43]. Previously, we used transgenic assays to dissect the sequences required for the sns5 insulator activity and defined nuclear factors that bind to these elements in homologous and heterologous systems [40,41,43]. Worth of mention, when placed in flanking location of a γ-retrovirus vector, sns5 prevents position effect variegation and improves transgene expression following vector integration in the erythroid cell genome [44]. Remarkably, sns5 favors the establishment of epigenetic marks positively correlated with the maintenance of a euchromatic state inside the integrated retroviral locus [44].
It is known that the expression of integrating vectors can be affected by several factors and, in some cases, insulator sequences could convey a fortuitous enhancer activity. Under this circumstance, an increase in transgene expression might not necessarily reflect a lack of variegation silencing.
In this study, we demonstrate that sns5 works as a chromatin insulator, counteracting position effects, when placed in flanking position of a green fluorescent protein (GFP) transgene contained in a lentivirus vector. Our results clearly indicate that the stimulation of transgene expression neither reflects an intrinsic sns5 enhancer activity nor relies on the recruitment of the erythroid-specific GATA-1 factor to sns5. Finally, in perfect accordance to the general belief that insulators shape the architecture of topologically independent chromosome domains [37,45–48], by using the Chromosome Conformation Capture (3C) technology, we report that a single copy of the aforesaid sns5-flanked lentivirus vector is specifically organized into an independent chromatin loop at the provirus locus.
Taken together, our results not only provide new clues concerning the molecular mechanism of sns5 function in the erythroid genome but also reassure the use of sns5 to improve the performance of gene therapy vectors.
Materials and Methods
Cell lines
Murine erythroleukemia (MEL)-58546, HeLa, and human 293T cell lines were grown at 37°C, in the presence of 5% CO2, in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM
Lentivirus vector constructs
The LV-GFP-sns5 vector series was generated by cloning, in sense orientation with respect to the viral transcription, either the wild-type 462 bp sns5 fragment or fragments bearing mutations in the GATA-1 binding sites located within the box-A and in both box-A and box-E (LV-GFP-sns5 mA and LV-GFP-sns5 mA+E, respectively), into the NheI restriction site of the 3′-LTR (long terminal repeat) of the pRRLSIN.cPPT.PGK.GFP.WPRE vector backbone [49]. The deleted construct used as positive control in the 3C assay was generated by digesting LV-GFP-sns5 with the ApaI restriction enzyme, followed by religation of the cut vector.
All the constructs were verified by restriction enzyme analysis and confirmed by DNA sequencing.
Vector production, vector titration, and MEL cells transduction
Vesicular stomatitis virus glycoprotein G-pseudotyped vector stocks were generated by a three-plasmid transfection of 293T cells. Viral titers were determined by transducing HeLa cells with serial dilution of the vector stocks and transduction efficiency was evaluated by quantitative polymerase chain reaction (qPCR) after 2 weeks of culture, as described [50].
1 × 105 MEL cells were transduced with either LV-GFP or LV-GFP-sns5 series vectors at multiplicity of infection (MOI) of 0.2–0.5, in the presence of 8 μg/mL polybrene. Sixteen hours after transduction, the cells were washed and plated in limiting dilution in 96-well flat-bottom dishes to grow individual clones. After 3 weeks of culture, GFP expression was assessed by flow cytometric analysis (FC-500 Beckman Coulter), and single-copy vector integrants were identified by qPCR analysis, as described [50]. Two independent transduction experiments were done for each construct. Although we did not experimentally confirm the genomic integration site for the single-copy integrants, we assume that the statistically relevant number of individual clones that we scored (>300 for each transduction experiment) most likely reflects independent integration events.
GFP expression of three to five clones for each construct, either silenced or expressed, was followed for a total of 8 weeks, showing unaltered expression over time. Furthermore, GFP expression status was faithfully maintained after freezing–thawing of transductants, definitively excluding any silencing effect.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) experiments were performed essentially as described previously [44,51]. Briefly, 3 × 107 MEL cells transduced with the LV-GFP vector series were crosslinked by incubating with 0.4% formaldehyde for 10 min at room temperature. Soluble chromatin complex in 1 mL of NICaCl2 buffer (15 mM Tris pH 7.5, 1 mM CaCl2, 60 mM KCl, 0.5 mM DTT, 15 mM NaCl, and 300 mM sucrose) was produced by digestion with 1.5 U/mL of micrococcal nuclease from Staphylococcus aureus (Sigma-Aldrich) for 7 min at 37°C. Under these conditions, the length of the digested chromatin ranged from 0.2 to 0.6 kb. The samples were incubated with 100 μL of salmon sperm DNA/protein-A sepharose slurry (Sigma-Aldrich) for 1 h at 4°C, with mixing. Twenty percent of chromatin was withdrawn (Input) and processed as the immunoprecipitated chromatin. Aliquots of chromatin containing 25 μg of DNA were incubated overnight at 4°C either in the absence of antibodies or with 5 μg of the anti-GATA-1 monoclonal antiserum purchased from Santa Cruz Biotechnology (Cat No. sc-265X). After extensive washing, the immune complexes were eluted with the elution buffer [1% sodium dodecyl sulfate (SDS), 0.1 M NaHCO3], and cross-links reversed by digestion with proteinase K at 65°C for 4 h. Five nanograms of the ChIPed DNA and input control, quantified by readings in a Qubit Fluorometer (Invitrogen), were used for qPCR analysis with Taqman chemistry and the following primers: sns5-forward: 5′-CCGGCA AATCAAGCTAAAGG-3′, sns5-reverse: 5′-AGGGCCGTTGAGGTGTTG-3′, sns5-probe: 5′-TGCACTCGCAAACC-3′.
Ct values obtained for each ChIP sample were normalized to Ct values of the input. Then, the percent of input values were calculated separately for each of the three replicates of an IP sample using the following formula: 2 × 100normalized Ct, and finally averaged.
Chromosome conformation capture assay and quantitative PCR analysis
Approximately 2 × 107 MEL cells bearing a single integrated copy of either the LV-GFP or LV-GFP-sns5 vector were crosslinked in 5 mL of 1 × phosphate-buffered saline (PBS) with 1.5% formaldehyde for 10 min at room temperature, with slow shaking. The use of single-copy transgenic lines avoided the possible complication of intercopy interactions. The crosslinking reaction was stopped by adding glycine to a final concentration of 125 mM, and the samples were kept on ice from this point forward. Cells were washed twice with ice-cold PBS by spinning down for 5 min at 3,000 rpm at 4°C in a Jouan BR 3.11 centrifuge. Cells were then resuspended in 5 mL of ice-cold cell lysis buffer (10 mM Tris pH 8.0, 10 mM NaCl, 0.2% NP40, 0.1 mM PMSF, 1.5 mg/mL pepstatin, and 10 mg/mL leupeptin), and incubated on ice for 1 h. Cell lysis was verified by microscope observations. After counting with a Burker chamber, 3 × 106 nuclei were washed twice with 0.5 mL of the appropriate 1.2 × restriction enzyme buffer (50 mM potassium acetate, 20 mM Tris-acetate pH 7.9, 10 mM magnesium acetate, and 100 μg/mL bovine serum albumin), and resuspended in 100 μL of the same 1.2 × restriction enzyme buffer. SDS was added to a final concentration of 0.3%, and nuclei were incubated at 37°C for 1 h, with slow shaking. Triton X-100 was then added to the final concentration of 1.8% to sequester SDS and samples incubated at 37°C for 1 h, with slow shaking. At this stage, a 150-μL aliquot of the sample was taken to be used as the undigested control.
Digestion was performed with 500 U of ApaI (New England Biolabs) at 37°C overnight, and the restriction enzyme was inactivated by the addition of SDS to 1.6% and incubation at 65°C for 20 min. At this stage, a 150-μL aliquot of the sample was taken to be used as the digested control.
The remaining reaction volume was diluted (to 2.5 ng/μL of genomic DNA) with a ligase buffer (50 mM Tris pH 7.5, 10 mM MgCl2, 10 mM DTT, and 1 mM ATP), Triton X-100 was added to 1%, and incubated for 1 h at 37°C. The DNA was ligated using 37.5 U/mL of T4 ligase for 4 h at 16°C. At this stage, a 150-μL aliquot of the sample was taken to be used as the ligation control.
Proteinase K was added at 20 μg/mL, and samples were incubated overnight at 65°C to reverse the crosslinks. The following day, samples were incubated for 30 min at 37°C with 0.5 μg/mL of RNase A, and the DNA was extensively purified by phenol extractions and ethanol precipitation.
Assessment of the ligated products was performed by conventional PCR, with primers designed so as to flank the religated ApaI sites. Thirty nanograms of DNA template were amplified in a 25 μL reaction volume using the following amplification program: 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 62°C for 30 s, and 72°C for 45 s. Specific ligation was confirmed by sequencing the obtained amplicons.
Primer sequences were as follows:
3C-for: 5′-GGAAAGAATAGTAGACATAATAG-3′;
3C-rev: 5′-AGGGAGCCGACTGCCGAC-3′.
Statistical analyses
All statistical analyses were performed with the STATA 12.0 software (StataCorp) applying either the Z-test (Figs. 1B, D and 2C, E) or the Student's t-test (Figs. 1C and 2D).

Expression analysis of the uninsulated and sns5-insulated LV-GFP vectors in MEL cells.
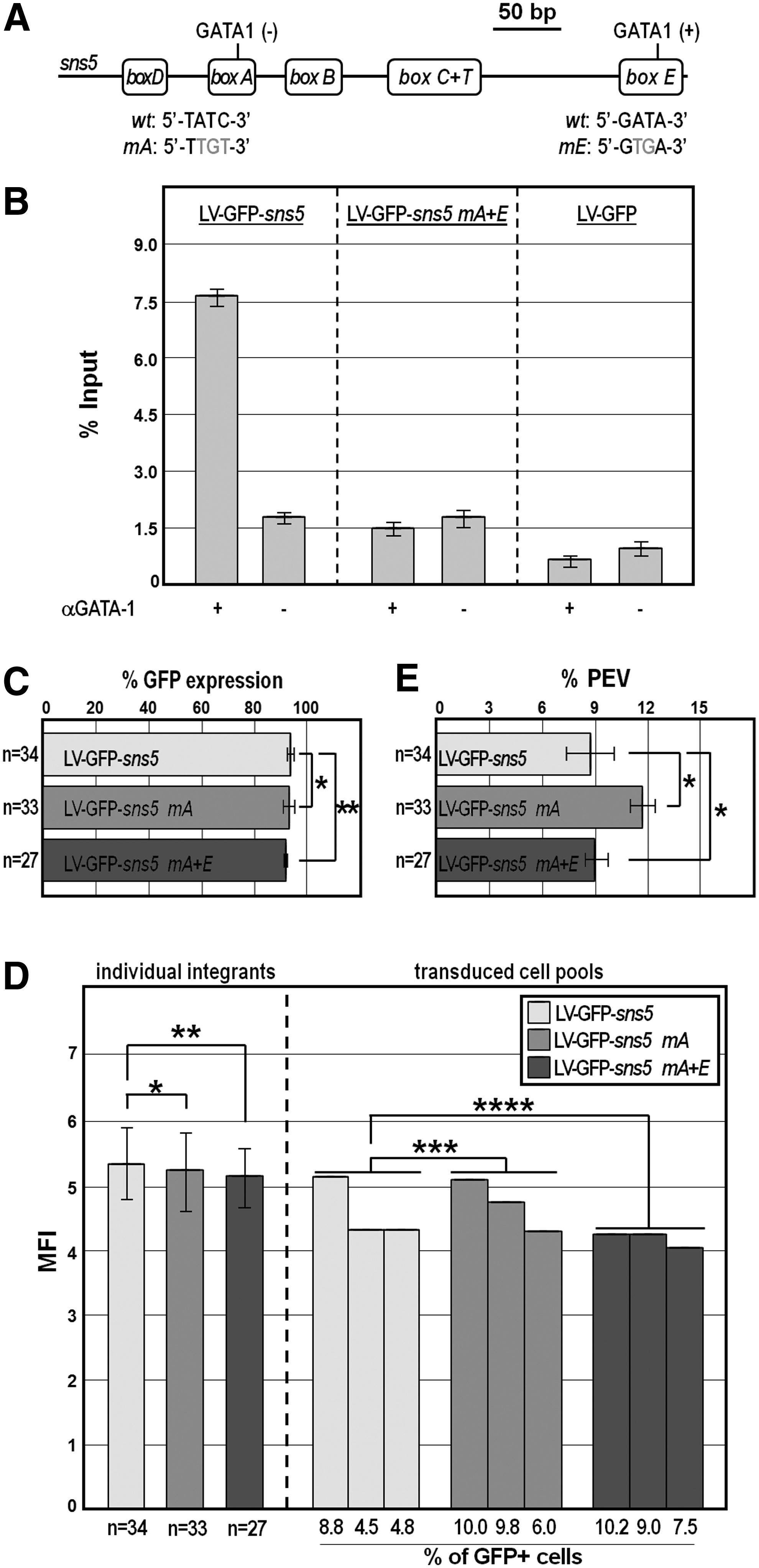
Mapping of GATA-1 binding on sns5 and expression analysis of LV-GFP-sns5 bearing mutated GATA-1 binding sites.
Results
The sns5 chromatin insulator increases the likelihood of transgene expression in a LV, minimizes the intraclonal variability, and has no enhancer activity
To test whether sns5 maintains the chromatin insulator properties in LV, a previously described LV expressing GFP from the pGK promoter (LV-GFP; Fig. 1A) was modified by cloning the 462 bp sns5 element in the 3′-LTR (in forward orientation with respect to the viral transcription) to obtain the LV-GFP-sns5 vector (Fig. 1A). Sequence analysis confirmed that the inserted sns5 fragment was faithfully copied and transferred into the 5′-LTR after the viral replication, entailing a double-copy arrangement of the sns5 element flanking the GFP transgene.
We then assessed the GFP transgene expression in single-integrant MEL clones following transduction with either LV-GFP or LV-GFP-sns5. MEL cells were plated at one cell per well to obtain a large panel of individual clones (n > 300 for each transduction experiment) that were simultaneously screened for both vector integration and GFP expression, by qPCR and flow cytometry, respectively. As shown in Fig. 1B, LV-GFP-sns5 provoked a strong increase in the fraction of GFP-expressing cells with respect to that resulted from the sns5-less vector (92% ± 2.3% vs. 43% ± 3.2%; P < 0.001). In principle, this result could confirm the insulator activity of sns5 in the lentivirus vector, unless an elusive enhancer activity specifically located in the sns5 sequence directly stimulated GFP expression. Data reported in Fig. 1C show that this was not the case, since the GFP mean fluorescence intensity of clones containing one integrated vector per genome was largely comparable between LV-GFP and LV-GFP-sns5-transduced cells (5.3 ± 1.6 and 5.3 ± 1.1, respectively; P > 0.75). We confirmed this result in a large panel of cells transduced with the viral supernatants at a modest MOI (see Materials and Methods section), to get less than 10% of transduced cells with a consequently limited vector copy number per cell (Fig. 1C).
Importantly, we also appraised that the intraclonal variegation of GFP expression, assessed as frequency of clones that showed silencing of GFP expression in the progeny, was significantly reduced in cell clones harboring the sns5-insulated vector (Fig. 1D). Indeed, about one-third of the clones transduced with the uninsulated vector exhibited variegation of expression (less than 100% of GFP-positive cells within the progeny) versus only ∼8% of clones harboring the insulated vector (P < 0.05).
Taken together, these results indicate that sns5 works as chromatin insulator in a lentivector and exclude any sns5-dependent enhancer activity.
The erythroid-specific GATA-1 factor is not involved in the insulator function of sns5
In a first attempt to explore the mechanism by which sns5 exerts insulator function in erythroid cells, we focused on the GATA-1 transcription factor. A recent interesting study described the interaction of GATA-1 to lentiviral provirus carrying the GATA1-HS2 erythroid-specific enhancer in both LTRs [52]. GATA-1 also recruits the CBP histone acetyltransferase, which in turn induces an open chromatin conformation throughout the provirus locus [52]. Worth mentioning, sns5 does contain two canonical WGATAR consensus sites, in the box-A and box-E regions, respectively (Fig. 2A), and both bind the GATA-1 factor present in erythroid nuclear extracts, according to gel shift experiments (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat) [43]. Furthermore, by ChIP analysis, we previously demonstrated the specific recruitment of GATA-1 on sns5 sequences in the active chromatin structure of integrated retroviral vectors [44].
To assess whether or not the insulator function of sns5 relies on GATA-1 binding, we, therefore, modified the LV-GFP-sns5 vector abolishing either the single GATA-1 binding site in the box-A (in the LV-GFP-sns5 mA vector) or both GATA-1 binding sites (in the LV-GFP-sns5 mA+E vector) by site-directed mutation (Fig. 2A). The resulting motifs were both incapable of binding to GATA-1, as highlighted by ChIP-qPCR experiments (Fig. 2B). On the other hand, transduction of MEL cells with LV-GFP-sns5 carrying either the wild type or the mutated version of the GATA-1 sites resulted in comparable transgene expression under the same experimental conditions described in the previous section (Fig. 2C–E), ruling out any involvement of the GATA-1 factor in the mechanism of sns5 boundary function.
Inclusion of sns5 in the LV-GFP vector mediates chromatin looping at the integration site
To investigate if chromatin looping could provide a mechanistic basis for the insulator activity of sns5, we applied the 3C assay to the LV-GFP provirus locus flanked or not by the sns5 element (Fig. 3A). 3C technique allows identification of physical contacts between DNA segments within the cell nucleus, and it has been successfully applied to analyze the three-dimensional architecture of chromatin domains within several loci [54–56]. MEL cells carrying a single integrated copy of either LV-GFP or LV-GFP-sns5 vector were exposed to formaldehyde, to capture the interactions of chromatin segments. The crosslinked chromatin and noncrosslinked genomic DNA template, used as a control, were then subjected to ApaI restriction. The positions of the two ApaI sites analyzed within the LV-GFP transgene are reported in Fig. 3A. The digested chromatin was religated at a low DNA concentration to favor intramolecular ligation. After the proteins were removed, interacting sequences were detected by conventional PCR. Primers for amplification were selected near the edges of the restriction fragment, so that the 3′-end of the primer turned to the fragment end (Fig. 3A).
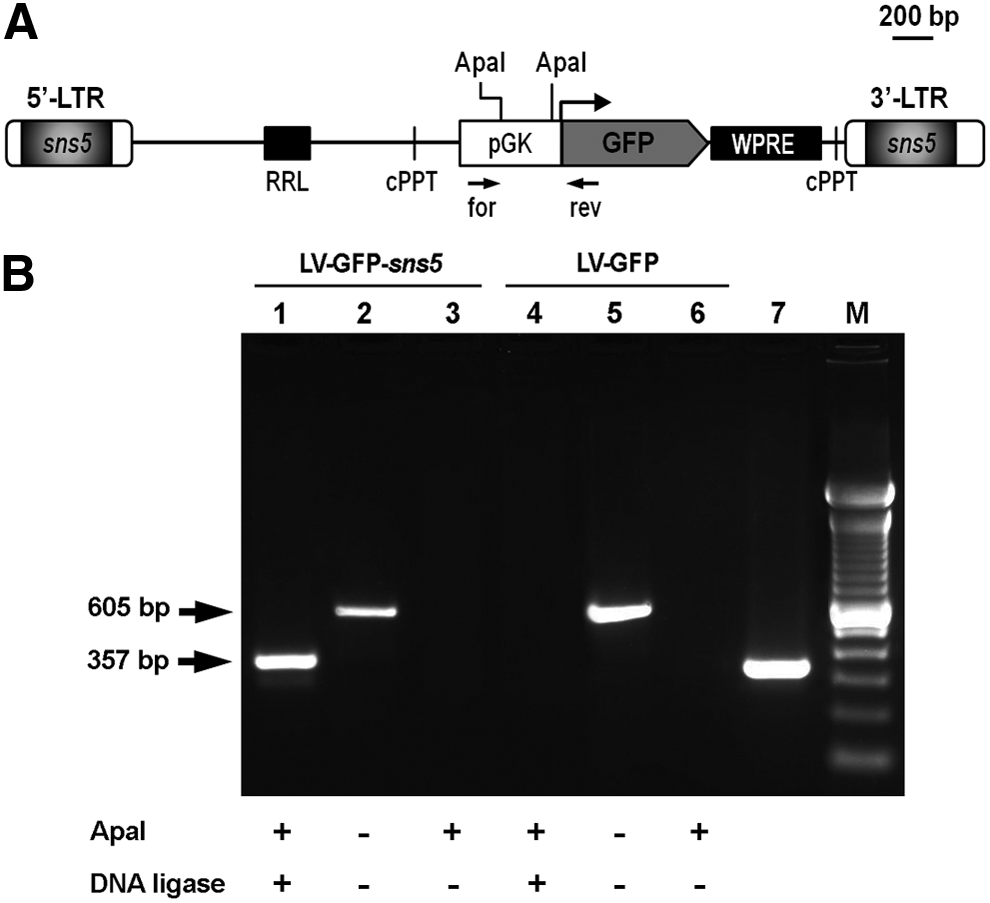
Chromosome Conformation Capture analysis of the sns5-insulated and uninsulated LV-GFP provirus locus.
As shown in Fig. 3B, the appropriate amplicon was generated when the chromatin DNA used was prepared from cells transduced with LV-GFP-sns5 (lane 1), but it was not obtained in any of the negative controls, viz the MEL cell genomic DNA and ApaI-cleaved, but not ligated chromatin DNA (lanes 2 and 3). The size of the PCR product was that predicted for the ligation of the juxtaposed cohesive ends of the two ApaI sites, and indeed, it coincided with that of the amplicon obtained from a deleted construct (devoid of the DNA fragment between the ApaI sites), used as a positive control (Fig. 3B, lane 7). The identity of the amplicons was eventually confirmed by DNA sequencing. Remarkably, no PCR product was detected in chromatin prepared from cells transduced with the uninsulated vector (Fig. 3B, lane 4). Altogether, these results strongly suggest that two sns5 insulator copies are located in close proximity to each other in vivo, and such a pairing likely mediates the formation of a looped conformation encompassing the entire provirus locus.
Discussion
The nature of the lentiviral infection cycle, in particular the random integration of the proviral form into the host genome, with a slight bias toward open and transcribed chromatin regions, provides desirable attributes that make LV vectors attractive for gene therapy approaches [53,57]. On the other hand, for the same mentioned reasons, LV vectors could induce the oncogene activity [58,59] and result in variegated expression, or even silencing, of their transgene cargo [60–62].
By virtue of the enhancer-blocking and/or barrier activities, chromatin insulators have been incorporated into LV gene therapy vectors to improve the performance of transgene expression and relieve the potentially adverse effects of random integration [63–66].
As a major drawback, it has been reported that the insertion of DNA fragments in the 3′-LTR provokes size-dependent detrimental effects on vector titers [67,68]. Accordingly, we previously demonstrated that incorporation of sns5 in γ-retroviral vectors did not affect the viral titer, likely due to the relatively small size of this element [44]. In addition, placing sns5 in flanking position of more complex LV vectors, containing larger functional cassettes bearing therapeutic units, did affect the viral titer at a very low extent (data not shown).
We have previously shown that the sea urchin-derived sns5 element maintains both the classical insulator activities in a γ-retroviral vector integrated in the erythroid cell genome [44]. In this work, we extend these findings, demonstrating that sns5 acts as a chromatin insulator when placed in flanking position of a GFP transgene contained in an LV vector. In particular, MEL cells bearing a single copy of the resulting vector exhibited a strong increase in the fraction of GFP expressing cells and reduced expression variability of the cell progeny, with respect to that resulted from the sns5-less vector.
Some recent successful gene therapy trials, as well as similar reports in mice and nonhuman primates, highlighted that the enhancer elements embedded in the integrated vectors could activate cellular oncogenes at a long distance, culminating in neoplastic transformation (reviewed in Refs. 13–15). In principle, this aspect could be adequately circumvented by the incorporation of chromatin insulators, assuming that these elements do not unexpectedly exert an intrinsic enhancer activity when placed in transgene constructs. Such a risk increases especially when heterologous insulators recruit transcription activators when conveyed in the human cell nucleus. This is exactly the case of the sea urchin sns5 insulator, which we have demonstrated previously to bind erythroid-specific and ubiquitous transcription activators, such as GATA-1, Oct-1, and Sp-1, in mammalian cells [43–44]. Nonetheless, our results clearly indicate that sns5 does not exhibit any enhancer activity in living erythroid cells. Although we did not analyze the specific expression of resident genes neighboring the vector integration site, our results make highly unlikely the propensity of the sns5-insulated integrating vector to perturb normal gene expression. Such a speculative deduction is further corroborated by previous findings demonstrating that EGFP transgene expression was barely detectable in human U2-OS cells stably transfected with either a plasmid or a retrovirus vector bearing the sea urchin insulator placed between the CMV enhancer and the tk promoter [42].
Heretofore, we also reported the association of GATA-1 and H3K4me3/H3K9ac-modified nucleosomes to sns5 [44], suggesting that this element is able to modify nucleosomal histones to maintain a euchromatic state inside the provirus locus. Other authors have also shown that GATA-1, bound at the GATA1-HS2 enhancer cloned in the lentiviral LTR, recruits the CBP histone acetyltransferase, which in turn favors the establishment of an open chromatin conformation throughout the provirus locus [52]. In our assays, LV-GFP-sns5 vectors mutated in the GATA-1 sites, and therefore unable to bind GATA-1, behaved as the wild-type vector when used in transduction of MEL cells. Thus, although GATA-1 is an important regulator of erythroid cell differentiation, and it is specifically bound to sns5, it is not involved in the mechanism of sns5 function.
Compelling insights on this issue came instead from 3C assays in samples derived from cells transduced with the LV-GFP-sns5 vector. These results strongly suggest that the two sns5-insulator copies flanking the provirus locus are located in close spatial proximity to each other in vivo. Stated in other words, the double-copy configuration of sns5 is required to organize the chromatin of the provirus locus into an independent topological domain. This finding, together with our previous functional results, leads to the most obvious interpretation that sns5 embeds the LV vector sequences into a chromatin loop that is protected from the encroachment of the host heterochromatin.
Taken together, our results not only confirm that sns5 represents a valid tool to shield integrated transgenes from negative chromosomal position effects but also suggest the mechanistic insight by which sns5 works within the erythroid cell nucleus. These aspects will be helpful in pursuing the improvement of the therapeutic potential of LV-mediated gene therapy approaches.
Footnotes
Acknowledgments
We thank Barbara Spina for technical assistance and Angela Vitrano for statistical analysis.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.