
Abstract
Registration of pharmaceuticals requires an assessment of their genotoxic potential using in vitro and in vivo tests outlined in the International Conference on Harmonisation (ICH) guidance S2(R1). We have evaluated numerous siRNA-N-acetylgalactosamine (GalNAc) conjugates containing phosphorothioate linkages and various combinations of 2′-fluoro and 2′-O-methyl ribose modifications of multiple nucleotides in the ICH battery of assays, all of which have uniformly yielded negative results. To verify these negative genotoxicity results, in this study we confirm test article exposure using toolkit small interfering RNAs (siRNAs) representative of those in the clinic. In the Ames test, the highest uptake of the siRNA-GalNAc conjugates occurred at 1 h postdose in all bacterial strains independent of siRNA sequence or chemistry (up to ∼14,000 siRNA molecules per cell), followed by metabolic degradation of the parent siRNA at 6, 24, and 48 h postdose. siRNA-GalNAc conjugates were internalized by bacteria as assessed by protection from the addition of nucleases to the culture media following uptake and by the requirement of cell lysis for detection of the siRNA. In the in vitro chromosome aberration assay, uptake was observed in Chinese hamster ovary cells (up to ∼5,500 siRNA molecules per cell at 21 h postdose) and in CD3+ human peripheral blood lymphocytes (up to ∼500 siRNA molecules per cell at 21 h postdose). In the in vivo micronucleus assay in rat bone marrow, exposure to parent siRNA was 100–350 μg of antisense strand per gram of protein at 24 and 48 h postlimit dose of 2 g/kg. Loss of terminal nucleotides was detected in bone marrow by mass spectrometry, indicating exposure to monomer metabolites as well. Negative genotoxicity results were also confirmed in an in vitro double-strand DNA break assay in HeLa and HepG2 cells where exposure was maximized using transfection reagents. Thus negative genotoxicity assay results for siRNA-GalNAc conjugates were valid and not the result of poor or no intracellular exposure.
Introduction
T
The bacterial reverse mutation (Ames) test [3,4] detects base-pair substitutions and frameshift mutations at the histidine or tryptophan locus in five auxotrophic gram-negative tester strains: Salmonella typhimurium TA98, TA100, TA1535, and TA1537 (or TA97 or TA97a), and Escherichia coli WP2 (or S. typhimurium TA102) [1]. The most widely used in vitro mammalian tests for chromosomal damage include the metaphase chromosome aberration assay, the micronucleus assay in human peripheral blood lymphocytes (hPBLs) or Chinese hamster ovary (CHO) cells, and the mouse lymphoma L5178Y cell tk (thymidine kinase) gene mutation assay (mouse lymphoma assay, MLA). In vivo test(s) are included in the test battery to account for absorption, distribution, metabolism, and excretion, and because some compounds are mutagenic in vivo, but not in vitro [1]. Chromosomal damage in rodent hematopoietic cells can be assessed using the micronucleus assay in erythrocytes (in blood or bone marrow) or the chromosome aberration assay in metaphase cells in bone marrow.
Oligonucleotide (ON) therapeutics have been uniformly negative in the standard test battery for genotoxicity across the chemical classes tested [5,6], including FDA-approved Macugen (pegaptanib) that contains both 2′-fluoro (2′-F) and 2′-O-methyl (2′-OMe) monomers, as well as phosphorothioate (PS)-substituted fomivirsen (Vitravene) and mipomersen (Kynamro). It is therefore consistent that siRNA-GalNAc conjugates that contain only these three chemical modifications are also negative in the standard battery (Ames, in vitro clastogenicity in hPBLs, and in vivo rat bone marrow micronucleus assay).
Both small molecules and ONs can have direct and/or indirect effects on DNA integrity. Potential indirect effects include downstream effects on DNA synthesis, cell cycle, or mitosis, all of which can be mechanistically similar between small molecules and ONs. However, potential direct effects on DNA integrity are quite distinct. Small molecules can modify DNA through covalent adducts, intercalation, cross-linking, and oxidative damage. In the case of ONs, nonnaturally occurring nucleoside monophosphates liberated by exo- and endonucleases theoretically can be converted to triphosphates by cellular kinases and incorporated into DNA, potentially leading to DNA replication or repair errors. If liberated in sufficient quantities, these monomers can also create an imbalance in endogenous nucleotide pools, which can result in chromosome breakage or mutation [7]. Nonnatural nucleotide triphosphates also have the potential to be incorporated into RNA, but this event would not be considered mutagenic because RNA is short-lived and the mutation would not be passed to progenitor cells. The third and most unlikely potential mechanism of ON genotoxicity is triplex formation, whereby an ON forms a triple helix with homologous DNA leading to site-specific mutations [8]. While the standard battery is designed to detect mutagenic incorporation and nucleotide imbalance, it cannot detect triplex formation between an ON and DNA because it is a sequence-specific event. However, ON-DNA triplex is unlikely to form at physiological salt concentrations even for ideal purine-rich sequences [9], and it is especially unlikely for siRNAs, which do not accumulate in the nucleus.
For small molecule drugs, passive diffusion into these test systems is assumed due to their small size (<1 kDa) and hydrophobicity—there is no requirement to demonstrate intracellular exposure. However, ONs are large (>5 kDa) and highly negatively charged, and thus passive uptake cannot be assumed. However, there are several reports documenting ON uptake. For example, free uptake of a single-stranded PS oligodeoxynucleotide has been assessed in S. typhimurium TA98, CHO cells, L5187Y mouse lymphoma cells, and mouse bone marrow [10]. Immunocytochemistry in the mammalian systems confirmed intracellular uptake, and the presence of metabolites suggested internalization by bacteria. In another example, activity of single-stranded antisense ONs (ASOs) and CpG ONs was detected in hPBLs [11], demonstrating intracellular uptake. In addition, one group showed intracellular functional uptake of unmodified siRNAs in Staphylococcus aureus [12]. However, to our knowledge, there is no evidence to date of intracellular uptake of double-stranded ONs into test systems of the standard battery and no direct confirmation of single- or double-stranded ON internalization in bacterial strains used for genotoxicity testing. Thus, confirmation of exposure in the test systems of the standard battery was warranted to verify negative genotoxicity results of siRNA-GalNAc conjugates.
Materials and Methods
Care and use of laboratory animals
All studies were conducted using protocols consistent with local, state, and federal regulations as applicable and approved by the Institutional Animal Care and Use Committees (IACUCs) at Alnylam Pharmaceuticals, Charles River Laboratories, or Covance, as applicable.
Good laboratory practice studies
Good laboratory practice (GLP)-compliant genetic toxicity studies were conducted using doses/concentrations up to the limit doses (except for ALN-AT3 in the rat bone marrow micronucleus test due to exaggerated pharmacology) with and without S9 metabolic activation as outlined in ICH S2(R1) and were all negative. In the bacterial reverse mutation assay, 1.58–5,000 μg/plate was tested in five bacterial strains: S. typhimurium TA1535 hisG46 rfa ΔuvrB, S. typhimurium TA1537 hisC3076 rfa ΔuvrB, S. typhimurium TA98 hisD3052 rfa ΔuvrB pKM101, S. typhimurium TA100 hisG46 rfa ΔuvrB pKM101, and E. coli WP2 trp uvrA. In the chromosome aberration assay in human peripheral blood lymphocytes, 1–500 μg/mL was tested in peripheral blood samples obtained from healthy, nonsmoking male donors, and stimulated with phytohemagglutinin M for 48 h before siRNA-GalNAc addition. In the in vivo micronucleus assay in bone marrow isolated from femurs, 500–2,000 mg/kg was tested by a single subcutaneous administration to young (47–56 days old) male Sprague-Dawley rats.
ON synthesis
Toolkit GalNAc-conjugated siRNAs mimicking those in the clinic were designed and synthesized by Alnylam Pharmaceuticals, as described by Nair et al. [13].
Bacterial cell culture
Fresh bacterial cultures of S. typhimurium STDisc TA98, STDisc TA100, STDisc TA1535, STDisc TA1537, and E. coli ECDisc WP2 (Molecular Toxicology) were grown in Nutrient Broth, Oxoid No. 2 (Molecular Toxicology) and were in the late-log phase of growth at the time of use. The density of the cultures was confirmed to be ≥1,000 × 106 bacteria/mL (OD600 > = 1.2).
Mammalian cell culture and transfection
CHO-K1 cells (ATCC) were cultured at 37°C, 5% CO2 in the F-12K medium (ATCC) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (pen/strep). Cells were seeded at 0.2 million/well in six-well plates for free uptake experiments. HeLa and HepG2 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and 1% pen/strep. Cells at 70% confluence were transfected with siRNAs or ASOs at specified concentrations using 4 μg/mL Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions. Fresh human whole blood (0.3 mL) was diluted in 4.5 mL culture media (RPMI 1640 supplemented with 1× HEPES, 2 mM
Rat bone marrow micronucleus assay exposure
Male Sprague-Dawley rats were administered an siRNA-GalNAc conjugate by a single subcutaneous injection to the upper back (500 and 1,000 mg/kg dose groups) or by two subcutaneous injections to the upper back at alternate sites (2,000 mg/kg dose group). Liver and bone marrow were collected at 24 and 48 h. Bone marrow was eluted from both femurs using Hanks' Balanced Salts Solution, cells were pelleted, washed thrice with PBS, and snap frozen in liquid nitrogen. The right lateral lobe of the liver was rinsed in cold saline and snap frozen in liquid nitrogen.
Stem-loop RT-qPCR
To quantitate exposure, cell pellets were resuspended in PBS containing 0.25% Triton X-100, heated at 95°C for 10 min, centrifuged at 14,000 rpm at 4°C for 10 min, and reverse transcription was performed on the supernatants using the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Briefly, lysates were denatured at 95°C for 10 min, and 5 μL was immediately added to 10 μL of ice-cold master mix containing 0.5 μM RT primer. Quantitative polymerase chain reaction (qPCR) was performed on Roche Light Cycler 480 II using LightCycler 480 Probes Master (Roche) according to the manufacturer's instructions. Primers (Integrated DNA Technologies) and probes (Thermo Fisher Scientific) are listed in Table 1.
siRNA, small interfering RNA; RT, reverse transcription; FWD, forward; REV, reverse; qPCR, quantitative polymerase chain reaction.
Protein quantitation
Protein content of lysates was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
Metabolic profiling
For the in vivo profiling of siRNA metabolites, bone marrow cells eluted from femurs using Hanks' Balanced Salts Solution were resuspended in the Lysis-Loading Buffer (Phenomenex) and extracted using the Clarity OTX SPE 96-well plate cartridges (Phenomenex). Approximately, 75 μL was subjected to LC/ESI-HRMS analysis using a Q Exactive mass spectrometer (Thermo Scientific). Data were processed using ProMass HR Deconvolution software (version 3.0, Novatia, LLC).
Serum treatment
Following incubations with siRNA-GalNAc conjugates (at concentrations and times specified in the figures), bacterial cell pellets were washed thrice with PBS, split into two tubes, and resuspended either in 50 μL PBS or 50 μL 50% human serum (Bioreclamation) diluted in PBS and supplemented with 10 mM magnesium chloride. After a 2-h incubation at 37°C, cells were pelleted at 3,000g for 10 min, washed thrice with PBS, and lysed for stem-loop reverse transcription-quantitative polymerase chain reaction (RT-qPCR) as described above. As a control for the serum nuclease activity, a free siRNA-GalNAc conjugate was incubated in PBS or 50% serum in parallel, and the reaction was quenched by the addition of 50 μL Clarity OTX Lysis-Loading Buffer (Phenomenex), followed phenol/chloroform extraction (Thermo Fisher Scientific).
RNA analysis
RNA was extracted with phenol/chloroform (Thermo Fisher Scientific), denatured at 95°C for 3 min in a formamide loading buffer, and resolved on a 15% denaturing TBE-urea gel (Thermo Fisher Scientific) in 0.5 × TBE. Gels were stained for 40 min with SYBR Gold (Thermo Fisher Scientific) diluted 1:10,000 in 0.5 × TBE.
Neutral comet assay
The neutral comet assay was performed using the CometAssay kit (TREVIGEN) according to the manufacturer's instructions. Approximately, 1,000 cells were plated per comet slide. DNA was stained using SYBR Gold (Thermo Fisher Scientific) and imaged using a fluorescent microscope (Zeiss AxioImager.Z1).
Results
Exposure to siRNA-GalNAc conjugates in the Ames assay
All siRNA-GalNAc conjugates tested to date were negative in the Ames assay (± S9 metabolic activation), when evaluated under GLP-compliant conditions (Table 2). To verify these negative results, we recapitulated standard Ames test conditions, in which up to 5 mg of an siRNA-GalNAc conjugate was added per plate (∼2 mg/mL before plating), followed by incubation in minimal glucose and minimal histidine/tryptophan conditions for 60–72 h. We did not use the S9 mix in our exposure experiments because the S9 does not activate siRNA-GalNAc conjugates, which are generally stable in S9 fractions with only minimal liberation of monomer metabolites due to the weak nuclease activity (unpublished data). Our aim was to quantitate exposure to parent drug using stem-loop RT-qPCR for the full-length antisense strand. Detection of intracellular parent drug suggests subsequent exposure to monomer metabolites and/or shortmer ONs.
All tests were performed in accordance with the OECD Principles of Good Laboratory Practice based on the OECD Guideline TG 473, and ICH Harmonised Tripartite Guidelines S2(R1) and M3(R2). All tests were negative.
Not at limit dose due to exaggerated pharmacology.
hPBL chrom abs, human peripheral blood lymphocyte chromosomal aberrations; ICH, International Conference on Harmonisation.
A nontargeting siRNA-GalNAc conjugate of the same chemistry as clinical candidates (containing PS, 2′-F, and 2′-OMe modifications) was incubated at 0.5 mg/mL with five bacterial strains routinely used in the Ames assay: S. typhimurium TA98, TA100, TA1535, and TA1537, and E. coli WP2 in liquid culture in nutrient-rich medium for 48 h (Fig. 1A). As expected based on data from GLP Ames studies, no cytotoxicity was observed. The parent antisense strand was detected in all strains: 0.2–40 ng per mL of lysate, which corresponds to 0.1–20 siRNA molecules per cell. The number of molecules per cell was determined using OD600 of 1.0 = 8 × 108 cells/mL, the volume of the culture, and the molecular weight of the antisense strand. Because the antisense strand was not detected in bacterial pellets when the cells were harvested and washed immediately after siRNA addition (t = 0 h), the siRNA was likely internalized rather than nonspecifically interacting with bacterial cell surface. Uptake was detected in both LPS-mutant (TA98, TA100, TA1535, TA1537) and LPS wild-type (WP2) strains, suggesting that the outer LPS layer does not limit siRNA uptake.
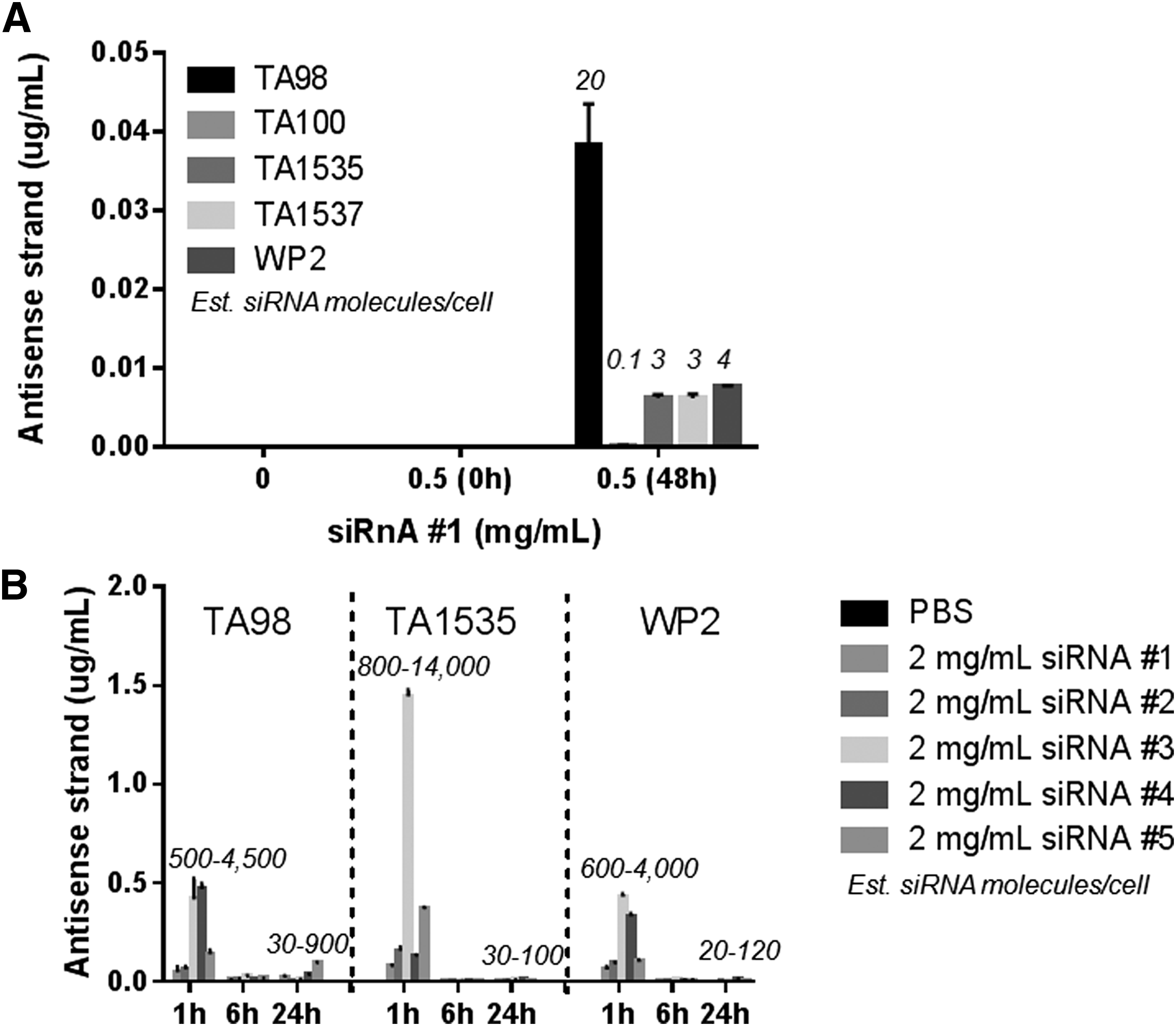
Chemistry- and sequence-independent free uptake of siRNA-GalNAc conjugates in the Ames test.
We hypothesized that the low abundance of the antisense strand at 48 h is due to metabolism of the parent siRNA by nuclease cleavage. We therefore selected three representative strains (TA98, TA1535, and WP2) for a time-course experiment with four different siRNA chemistries and two different sequences at a limit dose of 2 mg/mL (Fig. 1B). For all strains, chemistries, and sequences, the highest levels of the antisense strand were detected at 1 h postdose (up to ∼14,000 siRNA molecules per cell), followed by a significant decrease in signal at 6 and 24 h (93% decrease on average at both time points). Antisense levels at 24 h were ∼10-fold higher than at 48 h (compare Fig. 1A, B). Low efficiency of uptake past 1 h may be explained by the fact that the siRNA is unstable in bacterial supernatants (Supplementary Fig. S1; Supplementary Data are available online at http://www.liebertpub.com/nat): 85% of the parent antisense strand was lost after a 1-h incubation in supernatants from a 24-h TA98 culture, followed by a complete loss of the full-length signal at 6 and 24 h. These data suggest that rapid siRNA uptake at 1 h is significant and followed by metabolic degradation of the parent siRNA, indicating efficient liberation of monomer metabolites and/or shortmers.
The possibility remained that the siRNA was bound to the cell surface instead of being internalized to access bacterial DNA. To confirm intracellular uptake versus nonspecific cell surface binding, we incubated bacteria for 48 h with 0.5 mg/mL of the nontargeting siRNA-GalNAc conjugate and treated the resultant cell pellets with human serum in conditions that were optimized to achieve 90% loss of full-length free siRNA (Fig. 2A). Across all strains, only ∼30%–70% of the antisense strand was degraded following serum incubation (relative to PBS incubation), indicating that ∼20%–60% (90% baseline minus 70% and 90% baseline minus 30%) of the siRNA was protected from nucleases and thus likely internalized. Importantly, the nuclease activity of human serum was unaffected by the presence of bacteria and thus the decreased siRNA degradation in bacterial pellets was not due to the reduced nuclease activity (Supplementary Fig. S2A). Also, the serum nuclease activity was not internalized and was efficiently washed away as there was no change in endogenous bacterial RNAs (Supplementary Fig. S2B). In addition, bacterial cell lysis significantly increased stem-loop RT-qPCR detection of the antisense strand (5- to 30-fold depending on the strain), indicating that the siRNA is largely inaccessible to the RT enzyme in intact bacteria, and thus likely internalized after a 24-h incubation (Fig. 2B). A small amount of the antisense strand was detected without lysis (consistent with partial nuclease protection), and this fraction was lost with serum treatment before the RT reaction, confirming extracellular localization. In contrast, only 30%–50% of signal was lost when serum-treated bacteria were lysed before the RT reaction, indicating significant intracellular localization. There was no spontaneous lysis during serum incubation or the RT reaction (Supplementary Fig. S2C). Together, these data indicate that siRNA-GalNAc conjugates are internalized by bacterial strains used in the Ames assay.

Internalization of siRNA-GalNAc conjugates by Salmonella typhimurium and Escherichia coli.
Exposure to siRNA-GalNAc conjugates in the in vitro chromosome aberration assay
All siRNA-GalNAc conjugates were negative in the chromosome aberration assay in hPBLs when evaluated under GLP-compliant conditions (Table 2). To assess exposure, we mimicked hPBL assay conditions where human whole blood is first stimulated with the selective T-cell mitogen phytohemagglutinin M (PHA) for 48 h to promote proliferation of CD3+ cells, the siRNA is added at concentrations up to 500 μg/mL for 4 or 21 h, and chromosome aberrations are scored in metaphase-arrested dividing CD3+ cells. Following PHA stimulation, the nontargeting siRNA-GalNAc conjugate was added for 21 h, the cells were separated into the nondividing CD3− population and the dividing CD3+ population, and the antisense and sense strand levels and protein content (as a measure of cell number) were assessed (Fig. 3). As expected from GLP hPBL assay results, there was no cytotoxicity in either population up to the limit dose, and the uptake was dose dependent in both populations.

Exposure to an siRNA-GalNAc conjugate in the in vitro chromosome aberration assay. An siRNA-GalNAc conjugate was incubated with human peripheral blood lymphocytes (hPBLs) and Chinese hamster ovary (CHO) cells at indicated concentrations, and after 21 h, cells were harvested, exposure was assessed by stem-loop RT-qPCR for the sense and antisense and sense strand of the duplex, and normalized to the protein content of each sample. Human whole blood was stimulated with a selective T-cell mitogen (Phytohemagglutinin M) for 48 h before siRNA dosing, and the dividing CD3+ and the nondividing CD3− populations were separated. siRNA, small interfering RNA; RT-qPCR, reverse transcription-quantitative polymerase chain reaction.
In the dividing CD3+ cells, the uptake was ∼10-fold lower than in the nondividing CD3− cells: ∼3,500 versus ∼350 μg of siRNA per g of protein, or ∼5,000 versus ∼500 molecules/cell at 21 h after the limit dose of 500 μg/mL. The number of molecules per cell was determined by measuring protein content of known number of cells. The CD3−- cells are enriched in granulocytes, monocytes, and B cells, and thus the siRNA is likely taken up by phagocytosis in this population, potentially explaining higher exposure. These data show that there is significant exposure to siRNA-GalNAc conjugates in the hPBL assay in the dividing cell population where chromosomal aberrations are scored, and thus the negative genotoxicity results in this system are valid.
We also quantitated uptake in CHO cells, another commonly used in vitro chromosome aberration system (Fig. 3). Consistent with hPBL results, no cytotoxicity was observed up to the limit dose of 5 mg/mL, and the uptake was dose dependent. In CHO cells, the exposure after incubation with the nontargeting siRNA-GalNAc conjugate was approximately fivefold higher than in CD3+ cells (2,500 vs. 500 molecules/cell at 21 h after a dose of 500 μg/mL). Thus we confirmed dose-dependent free uptake of siRNA-GalNAc conjugates in both in vitro mammalian systems.
Exposure to siRNA-GalNAc conjugates in the rat bone marrow micronucleus assay
All siRNA-GalNAc conjugates were negative in the rat bone marrow micronucleus test when evaluated under GLP-compliant conditions (Table 2). To recapitulate the assay for exposure assessment, young male rats were administered a single subcutaneous dose of the nontargeting siRNA-GalNAc conjugate at 500, 1,000, and 2,000 mg/kg and bone marrow erythrocytes were collected from femurs at 24 or 48 h postdose. Exposure to the parent siRNA in bone marrow was dose dependent and reached an average of 250 μg antisense strand per gram of protein at 24 and 48 h postlimit dose as assessed by stem-loop RT-qPCR (Fig. 4A). As expected, exposure was higher in the target organ, liver, which expresses the asialoglycoprotein receptor (Asgpr) with high affinity for the GalNAc ligand. At the lowest dose of 500 mg/kg, liver exposure was 70-fold higher than bone marrow exposure at 24 h and that margin decreased to 40-fold at the highest dose of 2,000 mg/kg, suggesting Asgpr saturation and broader tissue distribution at the limit dose.

Exposure to an siRNA-GalNAc conjugate in the in vivo rat micronucleus assay.
Importantly, loss of terminal nucleotides was detected in the bone marrow by mass spectrometry, indicating exposure to monomers (Fig. 4B). At 48 h postlimit dose, 25% of the sense strand was lacking the 5′-terminal 2′-F-modified nucleotide, while 33% of the antisense strand was lacking the 3′-terminal 2′-OMe-modified nucleotide. The GalNAc ligand was also metabolized. Thus, bone marrow erythrocytes are likely exposed to parent drug and both 2′-F and 2′-OMe monomers in the rat micronucleus assay, thus validating the negative genotoxicity results in this system.
In vitro comet assay
Although exposure in all test systems of the standard battery for genotoxicity was confirmed, cellular uptake in these systems is limited to passive uptake, as the synthetic GalNAc ligand is designed to be taken up by the Asgpr on hepatocytes. As an additional exploratory measure, we transfected two nontargeting siRNAs with same sequence, but distinct chemistries into HeLa or HepG2 cells and explored an additional genotoxicity endpoint—the neutral comet assay for double-strand DNA breaks (Fig. 5A). Although a positive control, ISIS404130 [14], increased DNA breaks in both cell types after transfection, neither siRNA was genotoxic in this system as assessed by comet tail intensity and tail moment. As expected, transfection increased siRNA exposure 2–3 logs relative to the free uptake into an Asgpr-expressing cell line, HepG2, as assessed by RT-qPCR for the antisense strand (Fig. 5B). These results further confirm lack of genotoxicity of siRNA-GalNAc conjugates, even at increased exposure levels.
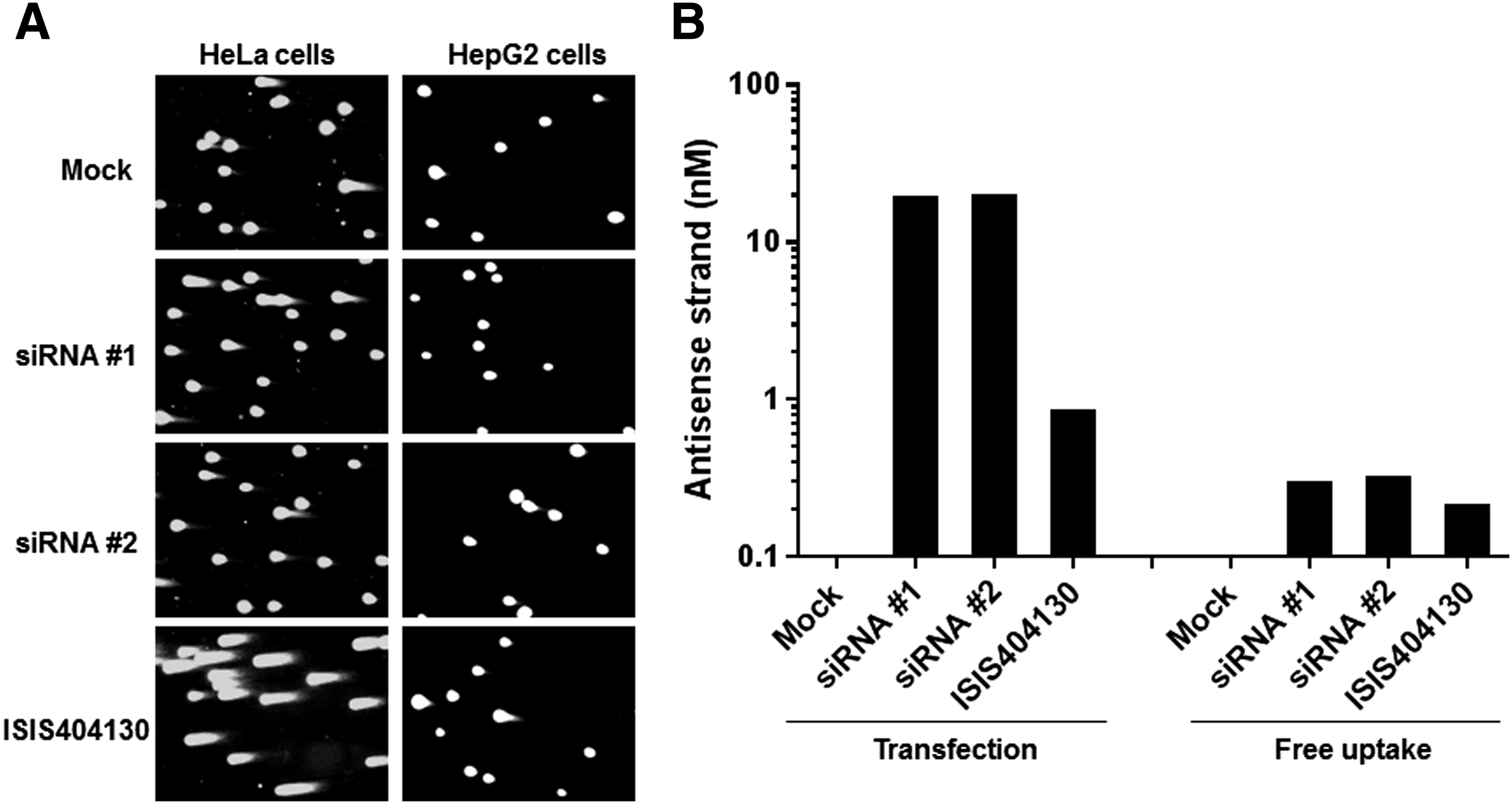
siRNAs are negative in the in vitro comet assay.
Discussion
siRNA-GalNAc conjugates containing 2′-F and 2′-OMe ribose modifications, as well as PS linkages, are not genotoxic, clastogenic, or aneuogenic as determined by the standard test battery for genotoxicity that includes a bacterial reverse mutations test, an in vitro chromosome aberration assay, and an in vivo micronucleus assay described in ICH S2(R1). In this study, we confirmed exposure in all three systems, thus verifying these negative genotoxicity outcomes. Although not a requirement in ICH S2(R1), understanding exposure provides valuable experimental context for siRNAs in these test systems. Lack of genotoxicity and free uptake into the test systems of the standard battery is consistent with previous reports for single-stranded unconjugated ONs for which intracellular uptake was demonstrated in CHO cells, L5187Y mouse lymphoma cells, mouse bone marrow [10], and hPBLs [11].
Gram-negative bacteria used in the Ames test are surrounded by a thin peptidoglycan layer sandwiched in between two lipid bilayers. Exposure to a full-length siRNA at 24 h appears to be lower in bacteria than in mammalian systems, either due to a lower uptake efficiency and/or higher intra- and/or extracellular nuclease activity. Despite the more stringent barrier, multiple lines of evidence support intracellular uptake of siRNA-GalNAc conjugates at high concentrations used in the Ames test (up to 2 mg/mL). First, no antisense strand was detected when cells were washed immediately after addition of the siRNA, arguing against nonspecific binding to the bacterial cell surface. Second, partial protection from nucleases was observed after incubation of an siRNA with bacterial cells. Third, only minimal amplification of the antisense strand was achieved in the absence of cell lysis. These data are consistent with a previous report of free siRNA uptake in bacteria, where 70% of the siRNA was in the cytoplasm and 30% in the membrane of S. aureus after a 4-h incubation [12].
It is possible that the siRNA is gradually embedded in the bacterial cell wall over time and thus inaccessible to added nucleases or the RT enzyme. Because we do observe metabolism of the parent siRNA, this would imply that there are endogenous nucleases present in the bacterial cell wall, which in fact has been reported in other Gram-negative species [15]. In this case, monomers liberated by periplasmic nucleases may easily diffuse through the single remaining lipid bilayer to reach the cytoplasm. However, there are also several possible mechanisms of intracellular uptake of nucleic acids into bacteria. For instance, the natural transformation process allows internalization of DNA and is documented in ∼80 bacterial species [16]. Although S. typhimurium and E. coli are not considered naturally competent, the genes mediating DNA uptake can be transcriptionally induced under certain conditions, including nutrient deprivation and other environmental stresses, and in some cases only a fraction of bacteria become competent [16,17]. In addition, there are various bacterial outer membrane secretory systems that mediate both active and passive uptake of solutes and macromolecules [18]. Indeed, there is precedence for functional siRNA uptake in Gram-positive bacteria encapsulated by a single lipid bilayer and a thick peptidoglycan layer [12].
Thus, negative results of the Ames test are likely not due to lack of exposure to siRNA-GalNAc conjugates. In fact, our data suggest that there is good exposure to full-length duplex and significant liberation of monomer and/or shortmer metabolites as the detection of the parent antisense strand is highest at 1 h and decreases 10-fold at 6 and 24 h, reaching only a few intact molecules per cell at 48 h. These findings are consistent with previously reported metabolite generation data for single-stranded PS oligodeoxynucleotide in S. typhimurium TA98 [10], despite the fact that siRNA-GalNAc conjugates are twice as large (∼16 kDa), have a less hydrophobic backbone (phosphodiester vs. PS), and are designed to be endocytosed by Asgpr. In addition, supernatants collected from a 24-h bacterial culture have a significant nuclease activity, leading to 85% reduction of the full-length antisense strand by 1 h (Supplementary Fig. S1), suggesting that monomers are generated rapidly in bacterial supernatants and can contribute to the intracellular pool acting on bacterial DNA following passive diffusion.
We also showed that siRNA-GalNAc conjugates are negative in the in vitro comet assay when exposure was maximized with transfection in HeLa or HepG2 cells. The most likely theoretical mechanism of ON genotoxicity is liberation of monomers (modified, nonnaturally occurring nucleotides) leading to incorporation into nuclear DNA during replication. As such, the in vitro comet assay conducted in dividing cell cultures should be a sensitive model. Option 2 of the ICH S2(R1) standard battery recommends the Ames test in combination with two in vivo assays: micronucleus or chromosome aberrations in bone marrow and, for example, an in vivo comet assay in the target organ. Because hepatocytes in a healthy liver rarely divide, monomer incorporation events are unlikely to be captured in the in vivo liver comet assay unless significant liver injury is triggered (regenerative response following insult). We are not aware of published examples of the comet assay applied to in vivo toxicology studies with ONs [6].
In conclusion, the weight of evidence presented in this study, including uniformly negative outcomes of GLP-compliant genotoxicity studies across all of Alnylam's programs to date and evidence of exposure in all test systems, suggests that siRNA-GalNAc conjugates and chemically modified monomers that comprise them are not genotoxic. Long-term rodent carcinogenicity studies with several siRNA-GalNAc conjugates have been initiated to further assess the risk of tumor-promoting effects.
Footnotes
Author Disclosure Statement
M.M.J., Y.J., R.G.D., A.N.H., J.L., and M.E.P. are employees of Alnylam Pharmaceuticals, Inc. The other authors have no competing financial interests to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.