
Abstract
The most widely used technique for the production of DNA aptamers/oligonucleotides is chemical synthesis. Despite its effectiveness, this technique cannot be performed “in house”, making the user fully dependent on a supplier. In this work, we present a simplified method by which it is possible to enzymatically produce DNA aptamers “in house”. This new method uses the rolling circle replication followed by a unique cleavage step using the SchI endonuclease. Potentially, any oligonucleotide can be produced by the enzymatic method proposed in this study. To illustrate, we present the production of three variations of the 31-TBA aptamer, a single stranded DNA which has anticoagulant action.
Introduction
S
To date, few methods describing the enzymatic production of ODNs have been reported [4–8]. In general, they are very creative and conceptually interesting, but their complexity (involving several steps, different enzymes and/or bacterial cultures, and diverse reaction conditions) and the absence of a simple method by which different oligonucleotides may be produced restrict the versatility expected for an “in-house” application. In this work, we present a simplified method based on rolling circle amplification (RCA) for producing ODNs.
Materials and Methods
The oligonucleotides used in this study were obtained commercially, and their purity was validated by PAGE. The T4 polynucleotide kinase (PNK) reactions were carried out using 2.5 μM of aptamer template, 50 mM Tris-HCl pH 7.6 at 25°C, 10 mM MgCl2, 5 mM DTT, 0.1 mM spermidine, 1 mM dATP, 0.5 U/μL T4 PNK (Thermo Scientific), and a final volume of 4 μL. The reactions were maintained at 37°C for 30 min and subsequently inactivated by heating at 75°C for 10 min. Ligation reactions were performed using the following: 1 μM of the DNA template; 60 mM Tris-HCl; 14 mM MgCl2; 12 mM DTT; 0.5 mM ATP; 10 μM of the O125R primer; and 0.1 Weiss U/μL of T4 DNA Ligase (Thermo Scientific); for 1.5 h at 22°C, increasing the final volume of the reaction to 10 μL. The reactions were inactivated by heating at 65°C for 10 min.
For enzymatic RCA, the following reagents were used: 0.5 μM of the circular template; 4.0 mM dNTP (1.0 mM each); 30 mM Tris-HCl; 33 mM Tris-acetate; 7.0 mM MgCl2; 10 mM Mg-acetate; 66 mM potassium acetate; 7.0 mM DTT; 0.1% Tween 20; and 0.2 U/μL phi29 DNA Polymerase (Thermo Scientific); adjusting the final volume to 20 μL. The reactions were incubated for 8 h at 30°C and then inactivated by heating for 10 min at 65°C. SchI digestion was carried out by adding 1 μL (10 U) of the enzyme into each reaction tube for at least 1 h; reactions were incubated at 37°C and then inactivated by heating for 20 min at 65°C. All reaction products were heated at 95°C for 3 min in a denaturing buffer (20 mM EDTA, 0.2% m/v bromophenol blue, and 80% formamide), resolved on a 8% PAGE (7 M urea, 40% v/v formamide), and were revealed using silver staining.
G-quadruplex structures and minimum free energy models were predicted using QGRS Mapper [9] and Vienna RNA Web Services [10] software, respectively; energy parameters for DNA were adopted in the latter [11]. DNA quantifications were performed using a NanoDrop2000 (Thermo Scientific) with 2 μL of each restriction product diluted 10× in ultrapure water. To investigate the effect of the structures presented by DNA templates, 220–320 nm absorbances were measured from solutions containing 10 pmol of each template in the reaction buffer without enzymes.
Results and Discussion
The method proposed in this study is based on the rolling circle replication of a circular DNA template and comprises four enzymatic steps as follows: (1) phosphorylation, (2) circularization, (3) polymerization, generating copies of the desired ODN in tandem, and (4) restriction assay (Fig. 1). Three different oligonucleotides were produced to validate the versatility of the method: 31-TBA aptamer, a blood coagulation inhibitor [12,13], and two variations of it; S31_TBA, a scrambled sequence, and T31_TBA in which guanines were replaced by adenines. Their respective molecular characterization, including the propensity to form secondary structures and G-quadruplexes, is shown in Table 1.
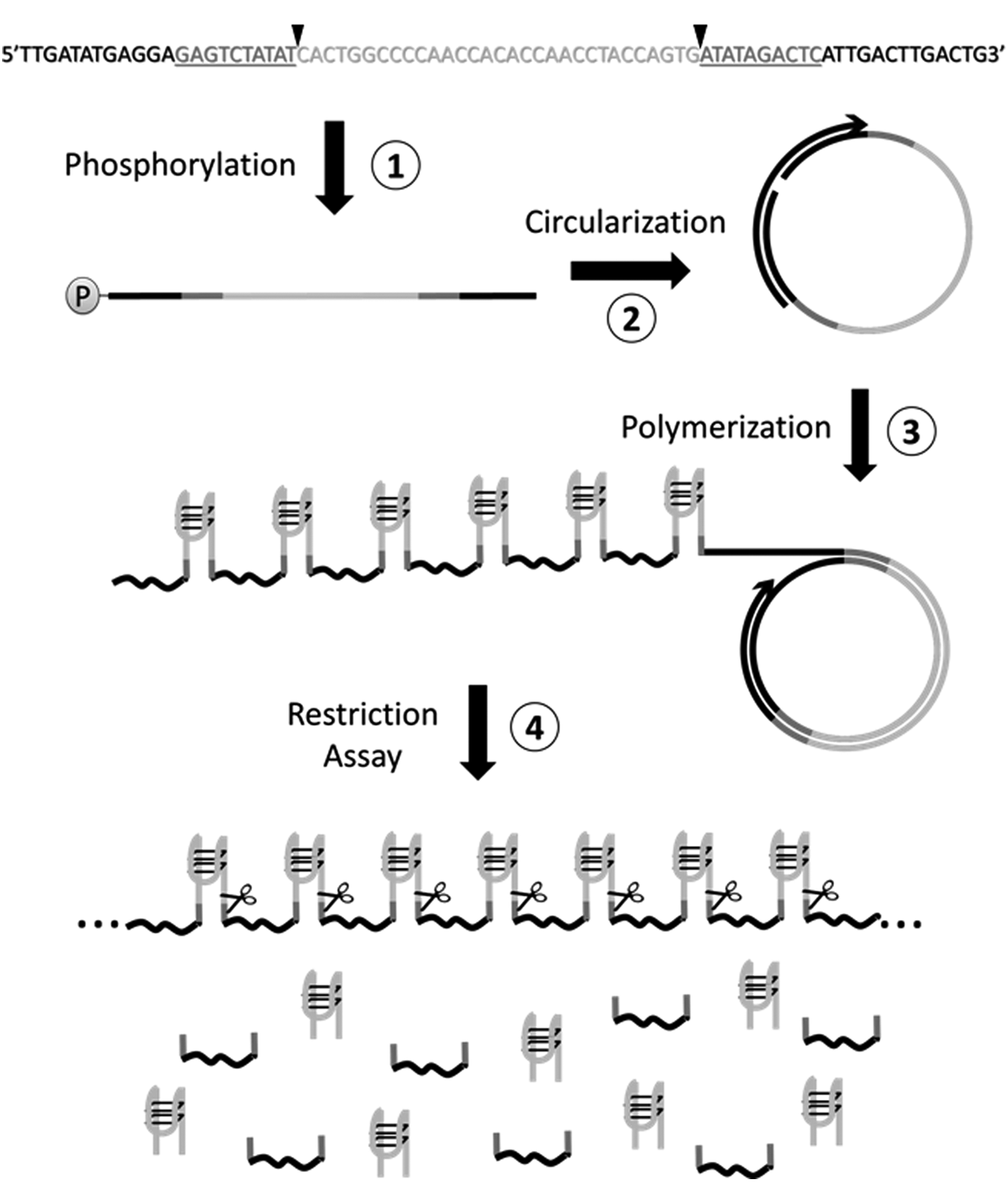
Schematic representation of the proposed method. The single stranded DNA template contains the reverse-complementary sequence of the desired aptamer (light gray), flanked by the SchI restriction sites (underlined). The black triangles demark the cut sites of the SchI endonuclease. The two sequences that form the primer annealing site should be at the ends of the DNA template (black). After the phosphorylation reaction (1), the DNA template was circularized (2), enabling the rolling circle replication (3). The amplicons contain tandem copies of the desired aptamer. The blunt end aptamers are released by digestion using the SchI endonuclease (4).
The G-quadruplex structure predictions were carried out according to Kikin et al. [9].
To produce 31_TBA aptamer (or any other desired ODN), its reverse-complement sequence should be designed at the central region of the DNA template (Fig. 1—light gray) and flanked by the two complementary sequences that form the SchI endonuclease restriction site (Fig. 1—underlined). The DNA template ends from the primer-annealing site after the ligation step (Fig. 1—black).
A ladder was then constructed using the DNA template, the 31-TBA aptamer, the O125R primer, two randomly-selected oligonucleotides, and another aptamer of 15 nt called HDI [13,14] (Table 1), to precisely determine the individual oligonucleotides generated after each enzymatic step. Furthermore, 5 μM of each oligonucleotide was subjected to a phosphorylation reaction to obtain a 5′-phosphorylated ladder. Profiles of the unphosphorylated and phosphorylated ladders are presented, respectively, in lanes 1 and 2 of Fig. 2. Phosphorylation causes an increase in electrophoretic mobility, which may be more easily observed in smaller oligonucleotides (Fig. 2—lane 2).
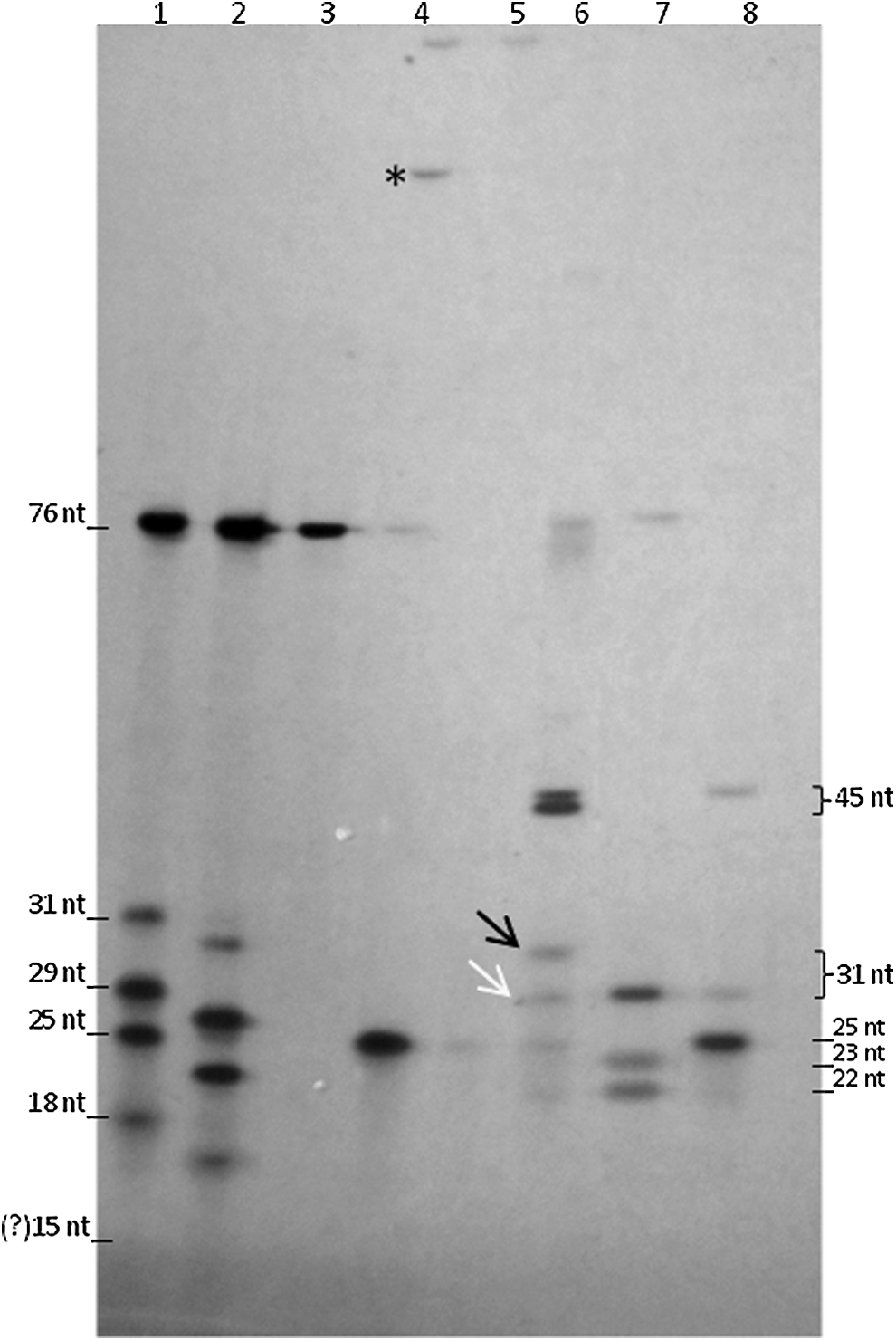
Expected fragments produced after each enzymatic step. The ladder containing the DNA template (76 nt), the 31-TBA aptamer (31 nt), three oligonucleotides (29, 25, and 18 nt), and the HDI aptamer (15 nt) was resolved before and after its phosphorylation (lanes 1 and 2, respectively). By comparing the profiles of the DNA template before and after the ligation reaction (lanes 3 and 4, respectively), it was possible to detect the band that corresponds to its relaxed circular form (lane 4, asterisk), confirming the efficiency of the ligation reaction. The RCA product could not enter into the gel mesh (lane 5). After SchI digestion, the 31-TBA aptamer (lane 6—black arrow) and the 45 nt oligonucleotide were detected. The complementary sequence of the 31-TBA aptamer could be identified (lane 6, white arrow) by comparing it to the profile of the digested DNA template (lane 7). Digestion of the circular DNA template released the 31-TBA complementary sequence (31 nt) and the 45 nt fragment corresponding to the primer annealing site fused to the restriction sequences (lane 8). The HD1 nucleotide (15 nt) could not be detected by silver staining. RCA, rolling circle amplification.
The four steps were successively performed in the same tube by the addition of each enzyme and its corresponding buffer. The DNA template was initially phosphorylated and subsequently circularized, forming the primer annealing site. The addition of the O125R primer in the reaction increased ligation efficiency due to the stabilization of the template ends. The template in relaxed circular form can be observed in Fig 2—lane 4 (asterisk). The largest band in lane 4 can be explained by the binding of two or more DNA templates, not affecting the next steps. As a control, the circular template was digested by SchI, generating the expected oligonucleotide of 45 nt, which contains the entire primer annealing site flanked by the restriction sites (Fig. 2—lane 8). Next, the RCA reaction was performed, producing large amplicons that did not enter into the gel mesh (Fig. 2—lane 5). After SchI digestion, the 45 nt oligonucleotides and the 31-TBA aptamers were released from the amplicon, confirming the effectiveness of the proposed method (Fig. 2—lane 6). The 31-TBA complementary sequence, a residue of the DNA template added in step 1, was also observed below the band corresponding to the aptamer. The 22 and 23 nt fragments observed in Fig. 2—lane 7 correspond to the restriction sites (10 nt) added to 12 or 13 nt of the primer annealing site.
The production of all expected fragments after each step confirms that the successive reactions occurred. However, the low intensity of the 31-TBA band (Fig. 2—lane 6) could indicate a loss in efficiency of some enzymatic steps. Interestingly, a careful inspection of the final reaction products revealed that the 45 nt product, which is equimolar to the aptamer produced, always presented a higher staining intensity. This interesting observation led us to investigate if the ODN structure could be influencing the staining intensity.
A serial dilution starting from 5 μM of each oligonucleotide used to mix the ladder revealed that the oligonucleotides that form G-quadruplex structures were stained less efficiently (Table 1 and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat). In agreement, the HDI aptamer could not be detected by silver staining, ethidium bromide, GelRed™ (Biotium), or HydraGreen™ (HydraGene), even using a concentration of 10 μM (data not shown). This result allowed for inferring that the 31-TBA aptamer is in a higher concentration than its complementary sequence, since the complementary sequence cannot form a G-quadruplex structure.
The same procedure was used for the production of S31_TBA and T31_TBA, and in both cases the yield of the reaction was much higher compared to the 31_TBA (Fig. 3A, B). The 260 nm absorbance measurements indicate that the yields of the S31_TBA and T31_TBA reactions were 41 μM and 50 μM, respectively, considering 100% reaction efficiency. These concentrations were estimated by subtracting the amounts of DNA template (0.5 μM) and primers (5 μM) from the total concentrations of each reaction (88 μM and 105 μM for S31_TBA and T31_TBA, respectively) and dividing the result by two. This division by two is necessary because the final product of each reaction comprises equimolar amounts of the oligo and its corresponding template.
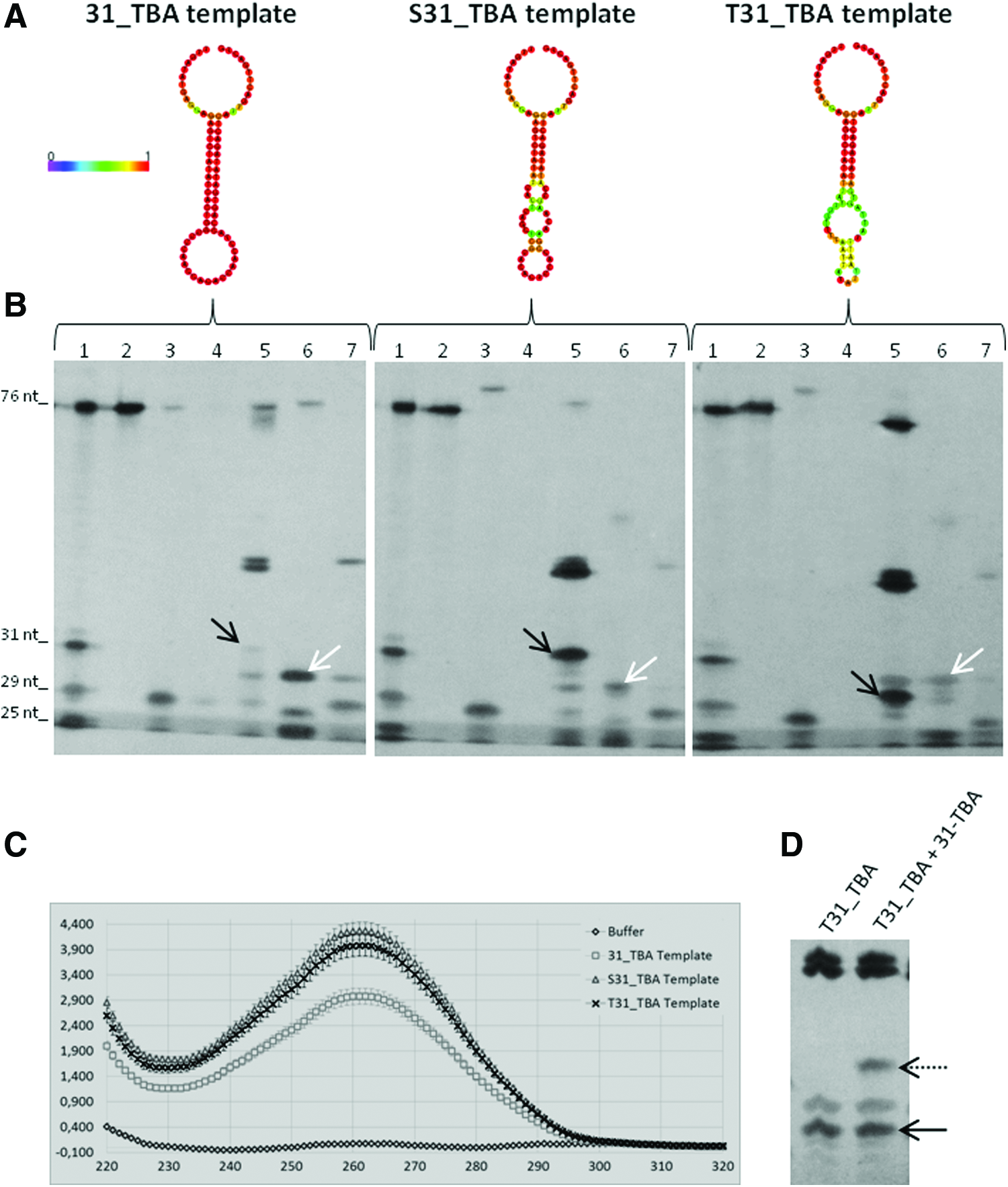
Propensity to formation of structures and its effects on RCA.
Due to the low yield of the 31_TBA reaction (Fig. 3B—left), a dilution curve shown in Supplementary Figure S1 was used to estimate the concentration of the 31_TBA aptamer. According to the intensity of the bands, the yield of the 31_TBA reaction is between 2.5 and 5 μM. The great difference comparing the yields of the three reactions can be explained by the propensity of the 31_TBA template to form stable structures. The minimum free energy models of each template (Fig. 3A) and their respective mountain plot representations (Supplementary Fig. S2) indicate that the stability of the 31_TBA template structure is significantly higher than the structures formed by the other templates.
The presence of structured regions (in which nitrogenous bases are not directly exposed) could also be confirmed by the lower absorbance at 260 nm observed in 31_TBA template solution compared with equimolar amounts of the other templates (Fig. 3C). The decrease in the yield of the reaction caused by the structured template cannot be minimized adding DMSO (in the proportions of 2.5%, 5%, or 10%) or betaine (0.75 or 1.5 M) in the reaction (data not shown). In addition, the presence of the 31_TBA aptamer at a concentration of 2 μM added before the reaction does not influence RCA, as confirmed by synthesizing the T31_TBA aptamer in the presence and absence of the 31_TBA aptamer (Fig. 3D). This test rejects possible inhibition or interference in enzymatic activity caused by the aptamer.
To conclude, enzymatic synthesis in the same tube proved to be efficient since all intermediary products and the desired aptamer were produced. The main advantages of this method are low cost for implantation and the versatility, as it can be implemented in any laboratory equipped with a water bath. Moreover, the minimization of stable secondary structures in the DNA template facilitates the chemical synthesis, the enzymatic processivity during RCA, and eliminates some potential sources of nonspecific amplification. Alternatively, the primer annealing site can be another oligonucleotide of interest. In this case, this method could be used to produce more than one ODN at the same time, in equimolar concentrations. This would facilitate, for example, the production of primer pairs required for PCR reactions or the production of complementary aptamers.
Footnotes
Acknowledgment
The authors thank the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for the financial support.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.