
Abstract
Uncontrolled bleeding is a major cause of mortality. Lysine analogues are routinely used in the management of bleeding, but several studies indicate a risk of serious detrimental effects upon their administration. In this study, we report a bivalent conjugate “3218” of two RNA aptamers selected for binding to the serine protease tissue-type plasminogen activator (tPA), the principal initiator of fibrinolysis in mammals. The constituent monomeric aptamers, K32v2 and K18v2, were previously demonstrated to weakly inhibit fibrinolysis. We now show that K32v2 and K18v2 recognize distinct binding sites, presumably in the A- and B-chain of tPA, respectively. Both aptamers bind tPA with low nanomolar affinity and inhibit tPA-mediated activities in a way that is consistent with the proposed localization of their binding sites. The 3218 conjugate possesses the inhibitory activities of both K32v2 and K18v2 and additionally exhibits increased inhibitory efficiency relative to the monomeric aptamers. The 3218 conjugate proved an efficient inhibitor of fibrinolysis and may find application in the management of bleeding as a substitute for, or in combination with, currently used lysine analogues.
Introduction
E
One possible way of managing traumatic bleeding is pharmacological inhibition of fibrinolysis [2]. Vascular injury induces blood coagulation, which serves to preserve the integrity of the circulatory system in the event of vessel rupture. The intent of therapeutics inhibiting fibrinolysis is the stabilization of fibrin aggregates that constitute the structural component of blood clots. In contrast to hemophilic patients, who lack blood coagulation factor VIII (hemophilia A) or IX (hemophilia B) and are treated by the administration of the deficient protein or activated factor VII [3], all trauma patients may benefit from inhibition of fibrinolysis to attenuate bleeding.
Following the discovery that severe adverse effects can accompany aprotinin administration [4], lysine analogues are the only approved medicinal products routinely used in the management of bleeding. Orally or intravenously administered trans-4-(aminomethyl)cyclohexanecarboxylic acid (tranexamic acid), a synthetic lysine derivative, has been used in the treatment of a wide variety of hemorrhagic conditions for more than four decades [2,5]. Tranexamic acid is generally considered to be well tolerated and notably has been shown not to increase the incidence of pathological thrombosis in clinical studies [6].
However, recent reports have suggested an increased risk of nonischemic seizure accompanying the administration of tranexamic acid before, during, and following cardiac surgery [7–10]. In addition, photoreceptor atrophy has been suggested as a consequence of prolonged use, and tranexamic acid is contraindicated in patients with defective color vision [11,12]. Furthermore, less serious adverse effects such as nausea and diarrhea have been reported as a result of tranexamic acid at high dosage [5]. Thus, these side effects can be problematic in patients with renal insufficiency [13,14]. In addition, cases of induced renal cortical necrosis in hemophilic patients as a result of tranexamic acid have been reported [15,16]. Finally, although rare, severe allergic reactions to tranexamic acid have been observed [17]. Hence, alternative antifibrinolytic agents might expand the availability of antifibrinolytic therapy to patients who are not eligible for the administration of tranexamic acid.
The serine protease tissue-type plasminogen activator (tPA) is the principal initiator of fibrinolysis in mammalian physiology. The vascular endothelium synthesizes and secretes tPA, and the rate of release is stimulated by the presence of procoagulant proteins (thrombin, fibrin, bradykinin, and so on) [18,19]. Once released, the protease binds to the surface of fibrin matrices through interactions with the finger and kringle 2 domains situated in the A-chain of tPA. tPA binds alongside plasminogen, a zymogen precursor to the serine protease plasmin, which is directly responsible for the resolubilization of fibrin aggregates [20,21]. The assembly of a trimeric activation complex at the fibrin surface stimulates the rate of tPA-mediated plasminogen activation ∼1,000-fold [22]. Lysine analogues such as tranexamic acid inhibit tPA-induced fibrinolysis by binding to the lysine binding sites on plasminogen and, presumably, tPA, thereby sterically hindering their association with fibrin. The relatively low affinity of tranexamic acid for plasminogen [23] necessitates administration at a relatively high dosage to achieve the desired effect.
Previously, we reported the isolation and characterization of two 2′-fluoropyrimidine-modified RNA aptamers selected for binding to tPA [24]. Nucleic acid aptamers, also known as chemical antibodies, are short single-stranded RNA or DNA oligomers (∼15–100 nucleotides) forming sequence-specific three-dimensional structures capable of recognizing a wide range of molecules, including proteins [25]. One (K18v2) was an efficient inhibitor of tPA-mediated plasminogen activation in the absence of soluble fibrin, while the other (K32v2) was not. Both aptamers, however, inhibited the stimulation of plasminogen activation conferred by fibrin but only inhibited fibrinolysis weakly in vitro. This observation would suggest that K18v2 has a binding site in the catalytic domain of tPA, while K32v2 is more likely to have an epitope distantly from the plasminogen recognition site, that is, in the A-chain. This hypothesis is not in contradiction to the ability of each of the two aptamers to interfere with fibrin stimulation of fibrinolysis, as mediated by the finger and kringle 2 domains [21], since RNA aptamers are, at least in some cases, able to influence competitive interactions of protein domains other than those with their primary binding sites [26].
Consequently, we have now sought additional evidence for the localization of their epitopes. We have done so to pursue the hypothesis that the two aptamers, which bind different epitopes, may be combined into a single molecule to yield a highly efficient inhibitor of fibrinolysis.
Materials and Methods
Materials
The tPA preparation used throughout this study was obtained from Boehringer Ingelheim and is identical to the preparation used clinically in fibrinolytic therapy. The purified catalytic domain of tPA was purchased from Sino Biological. Plasminogen was purified from human plasma on lysine Sepharose 4B resin (Sigma-Aldrich, St. Louis, Missouri) and eluted with six-aminohexanoic acid (Sigma-Aldrich). Human PAI-1 expressed in Escherichia coli was a kind gift from Agnieszka Jendroszek. Bovine fibrinogen was acquired from Enzyme Research Laboratories (South Bend, Indiana). Thrombin was a kind gift from John Fenton. Soluble fibrin was purchased from Aniara (West Chester, Ohio). Human plasma was obtained by depletion of erythrocytes and leukocytes (1300RCF, 15 min) from the blood of healthy donors collected in EDTA-coated Vacutainer® tubes (Becton, Dickinson and Company, Franklin Lakes, New Jersey). Triphosphate ribonucleic acids and deoxyribonucleic acids were purchased from Thermo Fisher Scientific (Waltham, Massachusetts). 2′-Fluoro modified deoxyribose pyrimidines were purchased from TriLink Biotechnologies (San Diego, California). Materials and reagents for surface plasmon resonance analysis were purchased from GE Healthcare (Uppsala, Sweden). Chromogenic substrates S-2251 (H-D-Val-Leu-Lys-pNA•2HCl) and S-2288 (H-D-Ile-Pro-Arg-pNA•2HCl) were purchased from Chromogenix (Milano, Italy). DNA oligonucleotide synthesis and plasmid sequencing services were provided by MWG Operon (Ebersberg, Germany). Visualizations of crystal structures were generated with the PyMOL Molecular Graphics System, Version 1.7.4 (Schrödinger, LLC. New York, New York). RNA secondary structures were predicted and visualized with the mfold web server [27].
RNA transcription and purification
Fluoropyrimidine-modified RNA was transcribed from dsDNA templates (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/nat) (1 pmol/μL transcription volume) by a T7 RNA polymerase carrying a Y639F mutation (produced in E. Coli with an N-terminal His-tag and purified by nickel column chromatography), which increases the incorporation efficiency of modified nucleotides.
Templates were generated as follows: complementary ssDNA oligos were complexed by heat denaturation (90°C 30 min) and slow renaturation (passive cooling to 37°C). The K18v2 and K32v2 sense–antisense pairs were used in RNA transcription at this stage, while the overlapping 3218 primer pair was subjected to Klenow extension before transcription. Klenow extension was accomplished by incubation with 50 units Klenow fragment without exonuclease activity (Thermo Fisher Scientific) and 320 nM dNTPs at 37°C for 90 min in a 500 μL volume. Extended pairs were purified by 6% native gel electrophoresis (loaded in 1:10 in 5.4 mL ddH2O; 3.9 mL glycerol; 500 μL 10% sodium dodecyl sulfate [SDS]; 200 μL 0.5 M EDTA), eluted in 0.3 M NaOAc, pH 5.2 at 4°C, and finally EtOH precipitated.
Transcriptions were carried out at 37°C for 14–16 h in 80 mM HEPES, 20 mM MgCl2, 1 mM spermidine supplemented with dithiothreitol and acetylated bovine serum albumin (BSA). Transcripts were purified by 8% (10% for transcripts <40 nucleotides) polyacrylamide gel electrophoresis, eluted in 300 mM NaOAc, pH 5.2 at 4°C, then EtOH precipitated and redissolved in ddH2O. Immediately before every experiment, RNA was retrieved from −20°C storage, thawed at ambient temperature, and then refolded in ddH2O by first denaturing secondary structure at 95°C, 10 min, then cooling to 20°C. The irrelevant control aptamer was the aptamer for urokinase-type plasminogen activator (uPA), upanap-12.33 [28].
Surface plasmon resonance analysis
Surface plasmon resonance measurements were carried out on a Biacore T200 instrument (GE Healthcare). For measuring the affinity of K18v2 to purified tPA, mAb clone HTPA2A153 was immobilized to ∼10,000 response units (RU) on a CM5 chip (GE Healthcare) by amine coupling chemistry. Then, purified tPA at 30 nM was captured to several hundred RUs on the active flow cell. Expressed wild-type or point mutants of tPA were captured from conditioned expression media and purified in situ by a sustained buffer flow. Both surfaces were then exposed to serial dilutions of K18v2. A reference recording from the upper flow cell was subtracted from the active cell recording, and the resultant adjusted sensorgrams were fitted to a 1:1 binding model with the Biacore Evaluation Software v. 1.0 (GE Healthcare). The sensor chip surface was regenerated by removal of bound RNA with an injection of HBS 0.1 M EDTA 0.5 M NaCl before injection of the subsequent sample. Complete regeneration, that is, dissociation of the tPA:mAb complex, was achieved with Gly-HCl, pH 2.7, following each complete dilution series.
To confirm the measured kinetic parameters by another format, a K18v2 variant with a 3′ purine extension (to ensure a non-fluor modified terminal nucleotide) was biotinylated by 3′ end ribose oxidation with sodium metaperiodate (Sigma-Aldrich) and subsequent reaction with EZ-Link Biotin-LC-Hydrazide according to the manufacturers' instructions (Thermo Scientific). Biotin-K18v2 was captured to tens of RUs on a CM5 chip coated with streptavidin, and dilutions of tPA were injected over the chip. Regeneration was accomplished by a 30 s injection of HBS with 0.1 M EDTA and 0.5 M NaCl. K32v2 was found to compete with mAb clone HTPA2A153 and was consequently only assessed in the biotin–streptavidin setup. Detection of binding to the catalytic domain of tPA was accomplished by direct immobilization by amine chemistry and subsequent injections of K18v2 and K32v2 at 150 nM.
PAI-1 inhibition of tPA
The relative rate with which PAI-1 inactivates tPA was estimated by the addition of 20 nM PAI-1 alongside 2 mM S2288 to an equal volume of equilibrated complexes of tPA (10 nM) and RNA (variable concentration). The absorbance at 405 nm was followed, and the values obtained at the conclusion of the tPA-PAI-1 reaction were plotted versus the corresponding RNA concentration.
For SDS-polyacrylamide gel electrophoresis (SDS-PAGE) analyses, 2 μg tPA was preincubated with or without a twofold molar excess of aptamer for 15 min at room temperature followed by a 2-min incubation at room temperature with 4 μg PAI-1 and S2288 to a final concentration of 1 mM. The reaction buffer was HBS pH 7.4 supplemented with 2 mM MgCl2.
Generation and expression of tPA alanine mutants
Point mutations were introduced to a pTT5 vector carrying the wild-type Homo sapiens tPA mRNA sequence (GenBank accession A07197.1) with the Phusion DNA polymerase (Thermo Fisher Scientific). Mutagenesis primers were designed using the online tool PrimerX (www.bioinformatics.org/primerx/) and acquired commercially (MWG Operon). Following an initial 2-min denaturation at 98°C, the following cycles were repeated 30 times: (i) 98°C 30 s; (ii) 60°C 60 s; (iii) 72°C 6 min, culminating in a final 72°C 10 min incubation to ensure complete extension. Remaining methylated bacterial DNA was digested with DpnI (FastDigest, Thermo Fisher Scientific), and extant plasmids were purified with the NucleoSpin PCR Cleanup kit (Qiagen, Venlo, Netherlands), then transferred into heat competent E. coli DH5α by a 42°C 45 s incubation, followed by 2 min on ice and 1 h of outgrowth in 2xTY media. Transformed bacteria were plated onto 2xTY agar supplemented with 1 μg/mL ampicillin and incubated for 16 h at 37°C. Five milliliter 2xTY media supplemented with 1 μg/mL ampicillin was inoculated with isolated colonies and grown overnight at 37°C in a shaking incubator, then purified using the NucleoSpin Plasmid kit (Qiagen) and validated by sequencing (MWG Operon).
HEK 293f suspension cells were maintained in Freestyle 293 Expression Medium (Cat. No. 12338-026; Thermo Fisher Scientific) under a 37°C 5% CO2 humid atmosphere in Corning Erlenmeyer flasks (Thermo Fisher Scientific). Cells were diluted with fresh media to a density of 0.2 × 106 cells/mL every 2–3 days and discarded after ∼30 passages. HEK293f cells were transiently transfected with plasmids produced in E. coli, using 1 μg DNA per mL of transfection volume, and linear polyethylenimine at a 3:1 ratio. Conditioned media were collected 5 days post-transfection by sedimenting suspended cells at 3,000RCF, then filtered through a 0.22 μm polyethersulfone syringe membrane. The tPA activity in conditioned media was quantified by the rate of S2288 (C = KM≈500 μM) hydrolysis and converted to an estimated concentration by comparison to the rate of hydrolysis by serial dilutions of purified tPA.
Plasminogen activation
The velocity of plasminogen activation by tPA was assessed by observing the rate of hydrolysis of S2251 at 500 μM in the presence of 2 nM tPA, 100 nM plasminogen, and a varying aptamer concentration in HBS supplemented with 2 mM MgCl2, 0.1 w/v% BSA, pH 7.5. Initial velocities were obtained by numerical integration of curve segments in the time range 10–30 min, where S2251 concentration and the rate of plasmin evolution are approximately constant. Resultant values are plotted against aptamer concentration and fitted to a sigmoidal model to obtain IC50 values. The stimulation of plasminogen activation exerted by a soluble fibrin fragment was assessed by the inclusion of 300 ng/mL soluble fibrin.
Fibrinolysis
Lysis of fibrin aggregates was assessed by incubating purified bovine fibrinogen (2 g/L) with thrombin (10 nM) in 100 μL HEPES-buffered saline supplemented with 2 mM MgCl2, 2 mM CaCl2, 1% BSA, pH 7.5 at 37°C in the wells of a Nunc microtiter plate alongside tPA (500 pM) and RNA (variable concentration) in a 200 μL volume. Exogenous plasminogen was not added since the fibrinogen sample was found to already contain this component. Clotting and lysis progress was measured by following solution turbidity as optical density at 405 nm.
Whole plasma clots were obtained by recalcification (10 mM CaCl2) of human plasma. Aptamer dilutions were allowed 15 min at 37°C to equilibrate with 200 pM tPA and 2 mM CaCl2 (final concentrations) in HEPES-buffered saline before addition of the same volume of undiluted human plasma.
Results
K18v2, but not K32v2, binds the catalytic domain of tPA
The proposal of a K18v2-binding site in the catalytic domain was evaluated by observing the binding of this aptamer to the isolated catalytic domain of tPA, as well as to the full length protease, by surface plasmon resonance analysis. While both aptamers bound immobilized full length tPA, only K18v2 produced a detectable signal upon injection over a surface coated with the isolated catalytic domain (Fig. 1). Thus, K18v2 binds the serine protease domain. K32v2 bound full length tPA, but not the catalytic domain, supporting the assumption that the aptamers bind separate sites in tPA and that K32v2 probably binds the N-terminal A-chain of tPA. An alternative explanation for the result could be that K32v2 only binds the catalytic domain conformation of the full length protein.

Analysis of binding of aptamers K18v2 and K32v2 to the catalytic domain of tPA. The purified catalytic domain of tPA was immobilized on a sensor chip surface by amine coupling chemistry, and the surface plasmon resonance response elicited by a 150 nM injection of K18v2 or K32v2 was recorded. A reference recording from a flow cell without immobilized protein was subtracted from the active cell recording, and the resultant sensorgrams are shown. tPA, tissue-type plasminogen activator.
The rate constants of the tPA-K18v2 interaction were determined by (i) the capture of tPA on a sensor chip coated with a monoclonal antibody directed at an epitope in the A-chain of tPA and the subsequent serial injection of decreasing dilutions of K18v2 (Supplementary Fig. S1A) or (ii) the injection of serial dilutions of tPA over a streptavidin-coated sensor chip surface with biotinylated K18v2 captured at low density (Supplementary Fig. S1B). The two approaches yielded comparable interaction kinetics. K18v2 bound tPA with a low nanomolar equilibrium dissociation constant, corresponding to relatively rapid rates of both association and dissociation (Table 1). Due to antibody competition and a high nonspecific binding, similar approaches could not be used for K32v2. However, the half-maximal inhibitory concentration for inhibition of binding of tPA to the immobilized antibody was 11 ± 3 nM (data not shown), suggesting a KD for K32v2-tPA binding in the low nM range. The observed competition of K32v2 with the A-chain binding antibody furthermore supports a binding site for this aptamer in the A-chain of tPA.
Rates of association and dissociation, as well as the equilibrium dissociation constant, for the interaction of K18v2 with purified, wild-type tPA, or tPA alanine point mutants, as measured by surface plasmon resonance (see Materials and Methods section). Kinetic parameters were determined by nonlinear regression analysis on the basis of a 1:1 binding model. Kinetic parameters of K32v2 could not be determined in the present setup.
tPA, tissue-type plasminogen activator.
K18v2, but not K32v2, inhibits tPA:PAI-1 complex formation
To map the K18v2 binding site on the catalytic domain of tPA, we first tested the effect of this aptamer on the reaction between tPA and its inhibitor PAI-1, which binds predominantly to the active site and the adjacent 37 and 60 loops [29]. The feasibility of this strategy was proven by our previous work with monoclonal antibodies against uPA [30].
To determine the effect of the aptamers on the binding of tPA to PAI-1, we utilized the fact that the aptamers do not inhibit the tPA-catalyzed hydrolysis of a small chromogenic substrate (data not shown), while PAI-1 does. We preincubated tPA with the relevant aptamers and added PAI-1 and a chromogenic substrate. The ability of the aptamers to inhibit the reaction of tPA with PAI-1 was quantitated from their ability to interfere with the inhibition of tPA-mediated turnover of the chromogenic substrate by PAI-1. Since the affinity of the available chromogenic substrate to tPA is too low to allow the determination of proper second order rate constants, we instead analyzed the degree of substrate hydrolysis upon complete inhibition of tPA by PAI-1 (Supplementary Fig. S2). While the reaction between PAI-1 and tPA was unaffected by an irrelevant aptamer and was complete in less than a minute, addition of K18v2 was able to delay the reaction between tPA and PAI-1 (Fig. 2). Hence, K18v2 binds tPA in a manner that inhibits PAI-1-mediated tPA inactivation. On the basis of this finding, we hypothesized that the K18v2 binding site is located at the “front” of the catalytic domain, where PAI-1 also binds. On the contrary, K32v2 had no measurable effect on the tPA-PAI-1 reaction, even at a concentration of 2 μM, an observation that is consistent with the assumption of the K32v2 binding site being different from the binding site of K18v2.
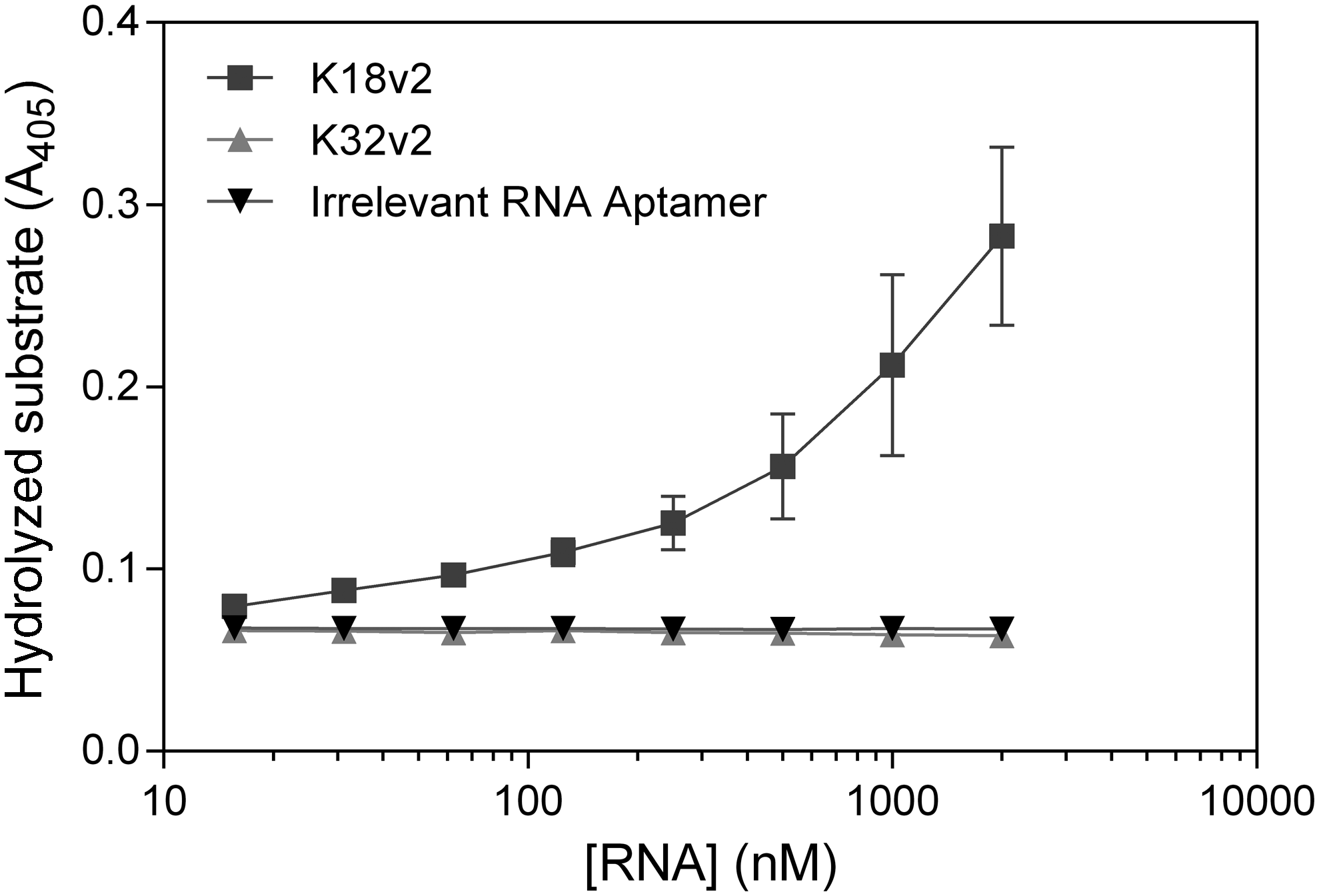
Inhibition of tPA:PAI-1 complex formation by K18v2. The relative efficacy by which K18v2, K32v2, and an irrelevant aptamer inhibit the reaction of tPA with PAI-1 was assessed from the total amount of hydrolyzed tPA-specific chromogenic substrate when the reaction of tPA with a twofold molar excess of PAI-1 was complete. The mean values and standard deviations from three independent determinations are shown. Representative raw data are given in Supplementary Fig. S2.
We also investigated the reaction between PAI-1 and tPA in the presence of the aptamers by SDS-PAGE analysis (Fig. 3). Upon preincubation of tPA with K18v2, K32v2, or an irrelevant aptamer followed by the addition of an excess of PAI-1, only K18v2 was able to inhibit the depletion of the tPA band and the formation of a band corresponding to the covalent tPA:PAI-1 complex. In another setup, the aptamers were added to already preformed tPA:PAI-1 complexes. As expected, none of the aptamers was able to reverse covalent tPA:PAI-1 complex formation once completed (data not shown).
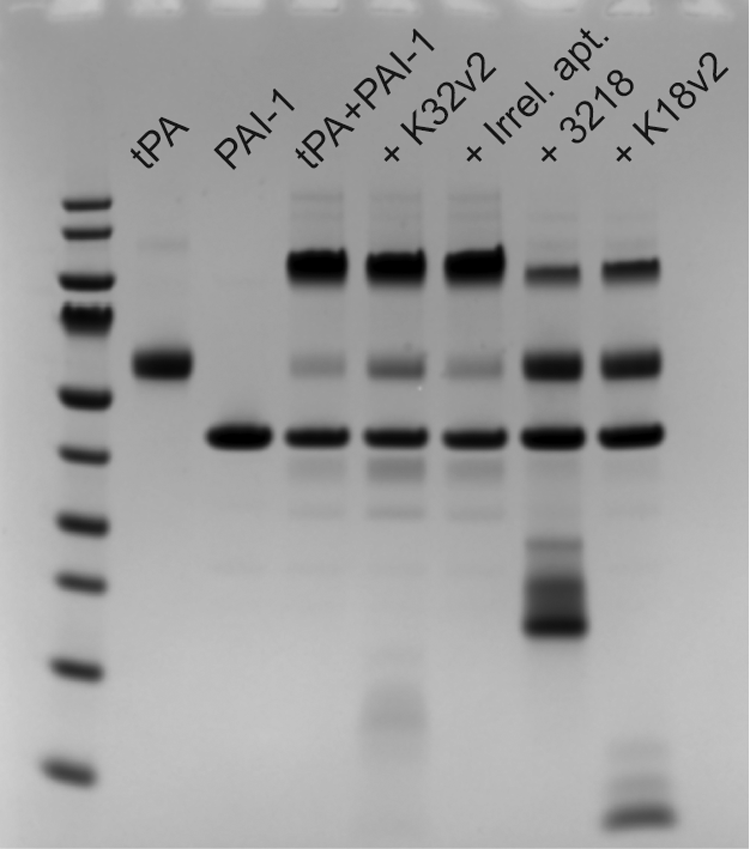
SDS-PAGE analysis of tPA:PAI-1 complex formation in the presence of aptamers. tPA was preincubated with the aptamers followed by the addition of an excess of PAI-1 relative to tPA. The mass (in kDa) of the protein ladder proteins: 180, 130, 100, 70, 55, 40, 35, 25, 15, and 10. Low molecular weight bands present in lanes with aptamers were also observed when aptamer samples were analyzed on their own (data not shown). SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
K18v2 engages tPA through residues in the 37 loop of the serine protease domain
Alanine scanning mutagenesis was used to delineate the binding determinants of the tPA-K18v2 interaction. As K18v2 is unable to bind murine tPA (data not shown), we compared the amino acid sequences of human and murine tPA to determine possible binding sites for the aptamer. One notable difference between murine and human tPA is the amino acid sequence of the 37-loop (KHRR in H. sapiens; KNKR in Mus musculus). Furthermore, the 37-loop and the 60-loop (ERFPPHHL in H. sapiens; ERFPPNHL in M. musculus) both participate in extensive contacts with PAI-1 [29], the binding of which is inhibited by K18v2. Both surface loops are located near the active site. Notably, K18v2 does not affect the rate of hydrolysis of small molecule substrates (data not shown). A binding site encompassing residues in either loop would likely shield the catalytic cleft and restrict the approach of macromolecules, such as plasminogen and PAI-1, while still permitting access by small molecule substrates.
To assess the effect of each substitution on the binding to K18v2, the tPA variants were captured to variable levels (hundreds of RUs) directly from dilute conditioned expression media by injection over a sensor chip surface coated with the A-chain-binding monoclonal antibody also used in the measurement of the tPA aptamer binding kinetics (Supplementary Fig. S3). Positions of interest were identified by screening for reduced binding to 75 nM K18v2, a concentration that produces a surface occupation of wild-type tPA in the dynamic range (Fig. 4A). The kinetic parameters of the interaction between K18v2 and variants that exhibited reduced binding were subsequently determined to quantify the reduction.

Kinetics of binding of K18v2 to tPA alanine mutants. Wild-type or alanine point mutants of tPA were captured to comparable levels (hundreds of response units) from conditioned expression media by a monoclonal antibody immobilized on a sensor chip surface (see Supplementary Fig. S3). Variants with reduced binding to K18v2 were identified by comparing the response to 75 nM aptamer
When normalizing the binding level of K18v2 for the tPA mutants to that of the wild-type protease, substantial and significant reductions (>50%, P < 0.05) were observed upon the introduction of alanine mutations at four positions: Lys36, His37, Arg37a, and Arg39 (chymotrypsinogen numbering). All four residues are situated in the 37-loop. Lys36, Arg37a, and Arg39 all exhibited ∼50% reduced steady-state binding to tPA, whereas the reduction was larger for the His37 alanine mutant. The Lys36 and Arg37a alanine mutants exhibited increased rates of dissociation and unchanged rates of association, whereas the opposite was true for the Arg39 mutant (Fig. 4B–F and Table 1). The altered interaction kinetics of these point mutant variants resulted in equilibrium dissociation constants of K18v2-tPA binding two- to threefold higher than those measured for wild-type tPA (Table 1). Injections of K18v2 dilutions over the His37 mutant produced binding signals that were much lower than those recorded for the wild-type, and the resultant curves were inadequate for data processing, indicating that tPA-K18v2 interaction is severely impacted by the absence of His37 (Fig. 4D). The residual binding observed was specific for the K18v2 sequence, as evidenced by the absence of any binding in response to the injection of an equivalent dilution series of an irrelevant RNA aptamer (data not shown).
To evaluate the structural integrity of the His37 mutant to ensure that reduced binding was not due to a distorted tPA conformation, the KM for S2288 hydrolysis by the single chain as well as the plasmin-activated mutant protease were determined and found to be very similar to that of the wild-type protease (Supplementary Fig. S4). Thus, His37 is crucial for the formation of a K18v2-tPA complex. Lys36, Arg37a, and Arg39 also contribute to K18v2 binding. The identified interacting and noninteracting positions are summarized in Supplementary Fig. S5.
Construction of a bivalent K32v2-K18v2 aptamer conjugate with improved inhibitory activity
We combined K18v2 and K32v2 RNA aptamers into a single construct, reasoning that a bivalent ligand encompassing two moieties with distinct binding sites might improve the affinity for tPA, relative to each monomeric constituent, to an extent enabling substantial competition with the avid tPA-fibrin association. The conjugate was produced by transcription from a synthetic dsDNA template constructed by Klenow extension of overlapping primer pairs encompassing, from the 5′ terminus, the sequence of K32v2, followed by an 18-nucleotide linker, and finally the sequence of K18v2 (Fig. 5). The linker sequence was designed to contain little or no secondary structure, as evaluated by in silico folding simulations, to avoid restricting the conformational flexibility of each constituent independently folding aptamer (Fig. 5). The conjugate construct was referred to as “3218.”
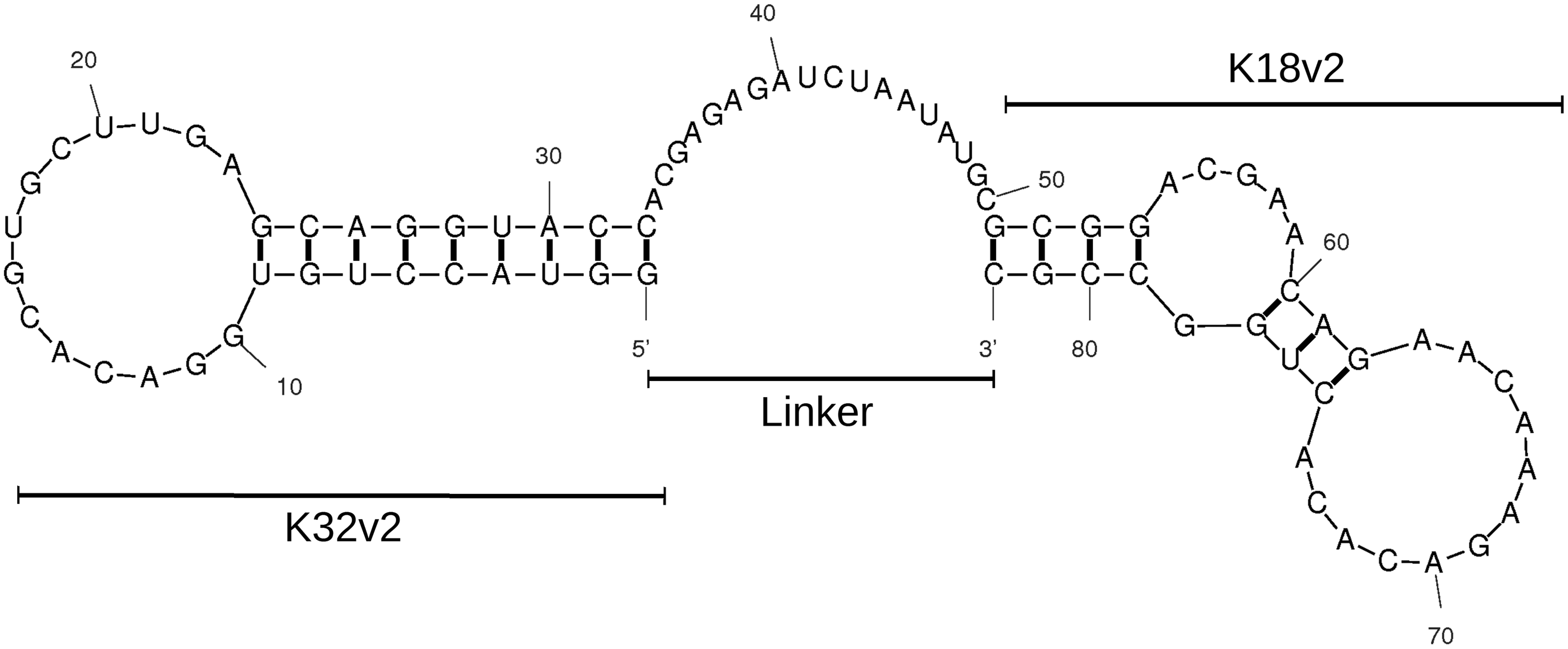
Predicted secondary structure of the K32v2-K18v2 (3218) conjugate. The sequences of K32v2 and K18v2 (individual MW ∼11 kDa), separated by an 18 nucleotide linker sequence (single-stranded region at position 33–50), combined into a single transcript (MW ∼28 kDa). Thick lines between letters indicate predicted canonical and wobble base pairing.
The activity of the construct relative to each monomeric constituent aptamer was evaluated by measuring the half-maximal concentration for inhibition of tPA-mediated plasminogen activation in the presence and absence of a soluble fibrin fragment (Fig. 6). Individually, K18v2 and K32v2 both inhibited the reaction in the presence of fibrin with half maximal inhibitory concentrations of ∼45 nM, while an irrelevant aptamer did not. The 3218 conjugate inhibited fibrin-stimulated plasminogen activation with half-maximal inhibition occurring at 1 nM, equal to half of the tPA concentration present (Fig. 6A). Hence, the 3218 conjugate inhibits fibrin-stimulated plasminogen activation at least 40-fold more efficiently than each constituent aptamer.
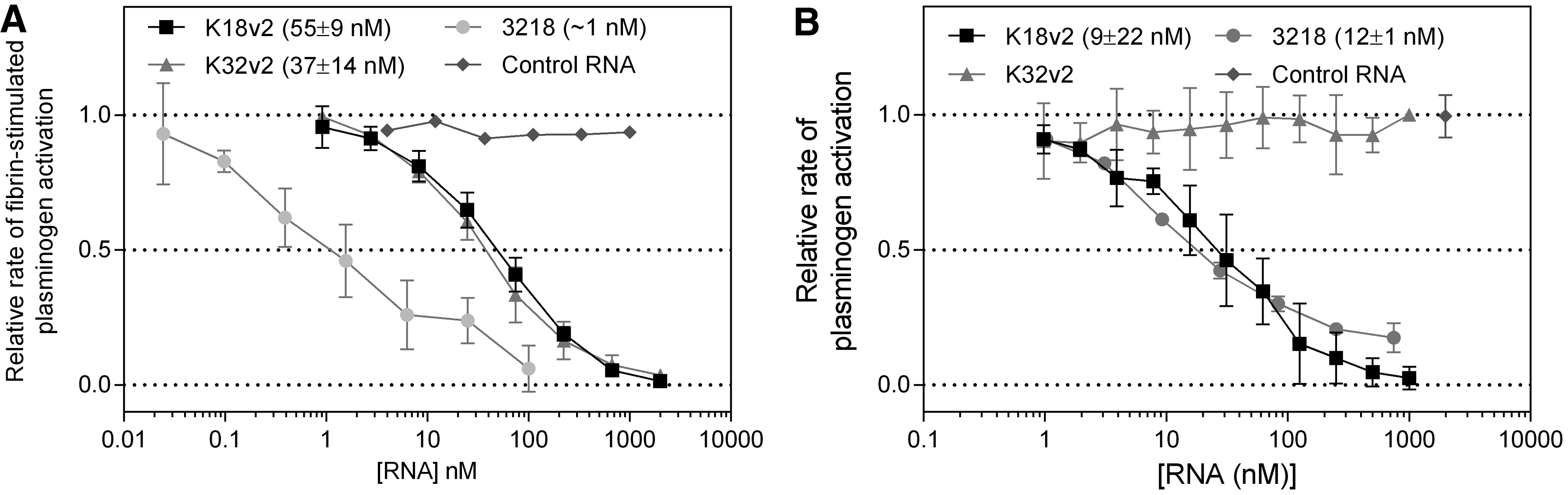
Effect of 3218 on tPA-mediated plasminogen activation. Aptamers were allowed 15 min at 37°C to equilibrate with tPA (2 nM) in the presence
On the contrary, the 3218 conjugate exerted an inhibitory effect on the activation of plasminogen in the absence of fibrin that was statistically indistinguishable from that of K18v2 alone (Fig. 6B).
Finally, the potency of 3218 to inhibit the reaction of tPA with PAI-1 was increased 100-fold relative to K18v2 and was saturable at ∼200 nM, although irreversible reaction of PAI-1 with tPA did eventually occur (Fig. 7). Interference with the tPA-PAI-1 reaction was not significantly different for K18v2 compared to the mixture of K18v2 and K32v2 (data not shown), demonstrating that the two aptamers must be covalently linked into the 3218 conjugate to exhibit the increased inhibitory activity. The ability of 3218 to interfere with the tPA-PAI-1 reaction was also confirmed by SDS-PAGE (Fig. 3).

Effect of 3218 on the PAI-1-mediated inactivation of tPA. Aptamers were allowed to equilibrate with tPA (5 nM) before the addition of PAI-1 (10 nM) and S2288 (1 mM). The total absorbance detected when substrate turnover had ceased is plotted versus the aptamer concentration. Data of monomeric K18v2 and K32v2 (from Fig. 2) are included to facilitate comparison. For an example of raw data, see Supplementary Fig. S2.
The 3218 conjugate inhibits fibrinolysis 100-fold more efficiently than monomeric K18v2 and K32v2
The potency of fibrinolytic inhibition by the 3218 conjugate was assessed in vitro and ex vivo by the lysis of aggregates prepared from purified fibrinogen and human plasma, respectively. The tPA-mediated resolubilization of pure fibrin aggregates proceeded readily upon application of 500 pM tPA, with half-maximal lysis occurring at ∼150 min (Fig. 8A). In the absence of tPA, however, no lysis was observed over the duration of the experiment (1,000 min). The inclusion of an equimolar 1 μM mixture of K18v2 and K32v2 delayed half-maximal lysis to ∼300 min, a twofold increase. An equivalent extension of lysis was achieved by the addition of the 3218 conjugate at a concentration approximately two orders of magnitude lower (12 nM), indicating an efficiency ∼100-fold higher than a noncovalent combination of each constituent aptamer. At the highest concentration tested (1 μM), the 3218 conjugate delayed clot lysis >6-fold to ∼1,000 min compared to uninhibited resolubilization.
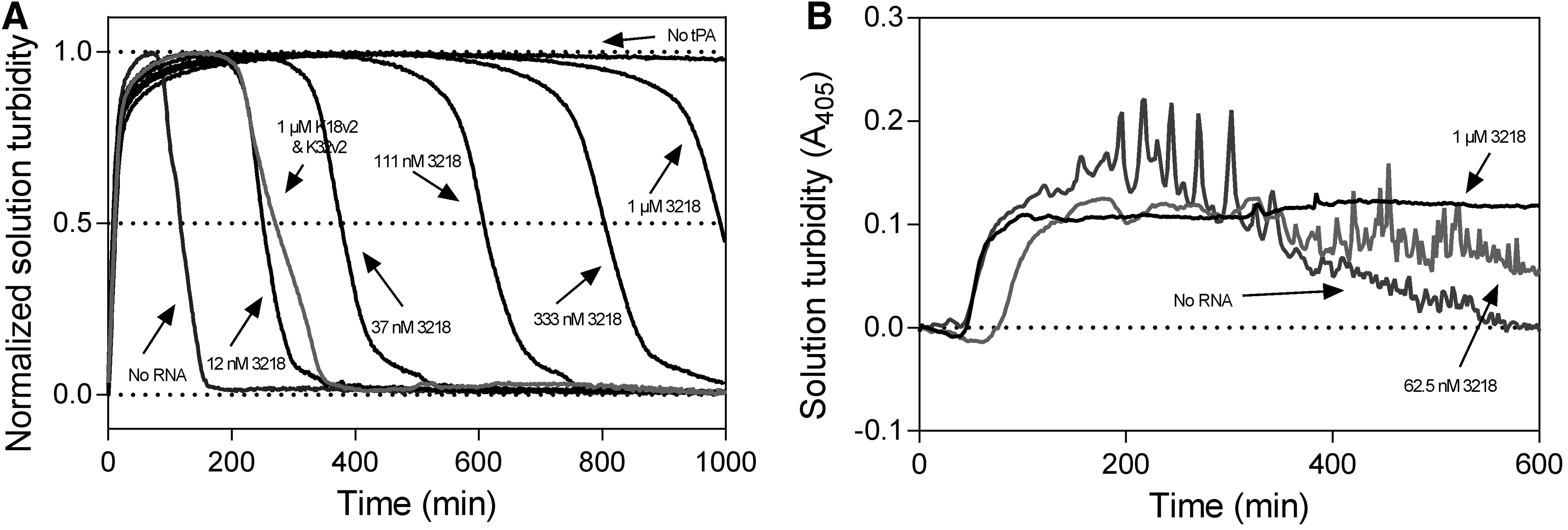
Effect of 3218 on the fibrinolytic activity of tPA. Fibrinolysis was assessed by
In an ex vivo clot lysis assay, in which an absence of inhibition by K18v2 and K32v2 was previously demonstrated [24], the 3218 conjugate was able to extend the lysis of whole plasma clots (Fig. 8B). At 1 μM, 3218 inhibited lysis completely for the duration of the experiment (600 min).
Discussion
In this study, we report that the aptamers K18v2 and K32v2 bind separate sites in tPA and most likely target the catalytic domain and the A-chain of tPA, respectively. The recognition of the catalytic domain by K18v2 was evident by the observed binding of K18v2 to the isolated catalytic domain, as well as from the reduced binding observed to variants of full length tPA with alanine mutations introduced in the 37-loop near the catalytic cleft. A-chain binding of K32v2 was reasoned from the lack of observable binding to the isolated catalytic domain, despite binding to the full length protease, and from competition of binding to tPA with a monoclonal antibody known to recognize an epitope in the A-chain of tPA.
The identified and inferred binding sites of each aptamer are consistent with their observed inhibitory activities, as summarized in Table 2. Both K18v2 and K32v2 evidently interfere with the fibrin-tPA association. Although the sites involved in fibrin binding are not precisely known, van Zonneveld et al. demonstrated an involvement of the kringle 2 and finger domains, while Bennett et al. identified positions in the catalytic domain that reduced fibrin stimulation upon alanine substitution [21,31]. In addition to fibrin binding, both aptamers hinder the binding of tPA to LRP-1 [24]. The importance for LRP-1 binding of a tyrosine residue in the epidermal growth factor domain was shown by Bassel-Duby et al. This observation is in agreement with the inhibition of the tPA-LRP-1 interaction exerted by K32v2 [32]. Understanding of the inhibition of LRP-1 binding by K18v2 is less straightforward. To our knowledge, no evidence in support of a direct interaction between the catalytic domain of tPA and LRP-1 is available. However, the related serine protease uPA, which shares 86% sequence identity with tPA, has been shown to bind LRP-1 through residues in the catalytic domain, in addition to residues in the A-chain [33].
n.a., not applicable.
Conjugation of K32v2 and K18v2 into a single construct resulted in a significantly increased potency of inhibitory activities exhibited by both moieties (fibrinolysis and fibrin-stimulated plasminogen activation). An increased potency was also found for the inhibition of the tPA-PAI-1 reaction, which is exclusive to K18v2. These findings presumably stem from the 3218 conjugate having an increased affinity for tPA compared to K18v2 or K32v2 separately due to the presence of two tPA targeting moieties in one molecule (ie, an avidity effect). The increased antifibrinolytic activity of 3218 confirms a previously stated hypothesis that inefficient inhibition of fibrinolysis by K18v2 and K32v2 is due to the affinities of the monomeric aptamers being insufficient for efficient competition with intact fibrin for binding to tPA [24].
On the contrary, inhibition of plasminogen activation in the absence of fibrin, an activity that is exhibited by K18v2 but not K32v2, was unaffected by conjugation. Considering the overlapping binding sites of plasminogen and PAI-1 in and around the catalytic cleft, this discrepancy is surprising. However, conjugation of ligands to the same protein has been observed to affect the potencies of each constituent moiety differently on at least one other occasion [34]. One possible explanation is that the kinetics of each separate interaction renders PAI-1 complexation more vulnerable to interference by a competitive ligand than plasminogen activation. If, for instance, the rate of Michaelis complex formation is substantially faster for tPA-plasminogen than tPA-PAI-1, the former interaction would be less sensitive to interference by a competitive ligand.
Nevertheless, the 3218 conjugate is a potent antifibrinolytic agent compared to monomeric K18v2 and K32v2, with possible applications in the management of bleeding due to excessive fibrinolysis. Although inhibition of tPA-PAI-1 reactivity is generally profibrinolytic, the magnitude of antifibrinolytic activity exerted by 3218 most likely negates any contribution by additional tPA inhibition. Adverse effects may be caused by the high dosage at which tranexamic acid must be administered to achieve sufficient antifibrinolytic activity. Plasminogen contains a number of lysine binding sites with affinities ranging from KD∼1,000 μM to KD = 1.1 μM [5]. The 3218 conjugate provides significant inhibition of fibrinolysis at concentrations 1,000-fold lower. Assuming similar pharmacokinetics and dynamics, a 1,000-fold dose reduction of the 3218 conjugate (relative to tranexamic acid dosage) might induce an equivalent magnitude of inhibition of fibrinolysis, but induce fewer or no dosage-related adverse effects. Moreover, the 3218 conjugate presumably inhibits fibrinolysis by the introduction of a steric conflict with tPA-fibrin binding. This mechanism is distinct from that of lysine analogues such as tranexamic acid, which inhibit the binding of plasminogen to fibrin by occupation of the lysine binding sites in its kringle domains. Hence, the 3218 conjugate is unlikely to interfere with the activity of tranexamic acid and vice versa. Consequently, superior antifibrinolytic activity may be achieved by coadministration of 3218 alongside tranexamic acid.
Footnotes
Acknowledgment
This work was supported financially by the Lundbeck Foundation (grant no. R83-A7828).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.