
Abstract
Activated protein C (APC) is a critical regulator of thrombin formation and thereby protects against thrombosis. On the other hand, overwhelming formation of APC increases the risk of bleeding such as in trauma-induced coagulopathy. Thus, pharmacological inhibition of APC activity may improve blood clottability in certain clinical situations. In this study, we demonstrate that the DNA aptamer HS02-52G binds with fast onset (1.118 ± 0.013 × 105 M−1 s−1) to APC and possesses a long residence time of 13.5 min within the aptamer-APC complex. Functional analysis revealed HS02-52G as a highly potent and specific inhibitor of APC in plasma and whole blood with IC50 values ≤30 nM, whose activity can be readily neutralized by the short complementary DNA molecule AD22. These features qualify the novel aptamer–antidote pair as a candidate treatment option for acute APC-related bleedings.
Introduction
H
An alternative supportive approach uses agents that downregulate endogenous anticoagulant mechanisms to restore the balance between pro- and anticoagulant processes. For instance, inhibition of tissue factor (TF) pathway inhibitor, an early regulator of blood clotting, has been shown to improve blood clotting in hemophilia [5].
A second major anticoagulant mechanism that can be modulated to improve blood coagulation is the protein-C (PC) pathway. This pathway is initiated when thrombin (that is released from the growing clot) forms a PC activation complex on the surface of luminal endothelial cells by binding to thrombomodulin. Once activated, the serine protease-activated protein C (APC) proteolytically degrades the activated coagulation factors, V (FVa) and VIII (FVIIIa), thereby downregulating the catalytic activity of the tenase complex and the prothrombinase complex. As a consequence, the rate of thrombin formation is significantly reduced and blood clotting effectively inhibited [6].
This mode of action explains that excess formation of APC might induce a hemorrhagic phenotype. It has been reported that overwhelming APC formation may play an important role in the pathogenesis of trauma-induced coagulopathy (TIC) [7–9]. TIC occurs in up to 45% of patients with severe trauma and significantly contributes to the morbidity and mortality of these patients [10]. Hence, inhibition of APC could be helpful in the prevention and treatment of TIC.
On the other hand, it has also been shown that the bleeding phenotype in patients with hemophilia is significantly reduced in the concomitant presence of the FV Leiden mutation, the major cause of hereditary APC resistance as a result of impaired cleavage of mutated FV(a) by APC [11]. Consequently, due to the obvious support of in vivo thrombin generation by impaired APC activity, APC has been recognized as a candidate target for the adjuvant treatment of hemophilia [12].
Thus, drugs that inhibit the anticoagulant activity of APC might be useful for the treatment of acute or chronic bleeding complications. Indeed, the design and evaluation of peptide and low-molecular-weight compound-based APC inhibitors have been described in the literature [13–17]. However, insufficient target selectivity and/or potential toxicity may be an issue of at least some of these molecules [13]. Furthermore, although the therapeutic index (safety window) of an APC-inhibiting drug may be considerably low in clinical practice, no antidote strategy to reverse the activity of such inhibitors in case of thrombotic complications has been reported so far. Moreover, in addition to its anticoagulant function, APC possesses cytoprotective and anti-inflammatory activities that are important for prevention of severe organ failure and that support wound healing of severely injured patients [18]. Thus, it is of primary importance to have available a controllable APC inhibitor.
We previously described the generation and characterization of an APC-targeting aptamer, HS02-52G [19]. By use of filter retention analysis, it has been demonstrated that HS02-52G binds with high affinity to both plasma-derived and recombinant APC [dissociation constant (KD) <1 nM], while binding to the zymogen PC was found to be on an ∼200-fold lower level. Furthermore, no binding of HS02-52G to the structurally related serine proteases, thrombin, activated factor VII, activated factor IX, and activated factor X, each tested at supraphysiological concentrations of up to 320 nM, was observed [19]. These characteristics make HS02-52G an interesting candidate molecule for the development of a fast-acting and well-controllable APC-inhibiting strategy.
In this study, we analyzed the impact of HS02-52G on the restoration of blood clotting in APC-affected plasma and whole-blood coagulation models. In addition, a series of designed HS02-52G-neutralizing antidote molecules has been investigated. The results reveal HS02-52G as an effective APC inhibitor whose activity can be readily inhibited by the antidote molecule AD22. The data demonstrate that this novel inhibitor–antidote pair is a potent and safe candidate for the development of an APC-inhibiting drug.
Materials and Methods
In silico analysis of DNA intramolecular foldings and duplex characteristics
The DNA folding form on the mfold web server available at http://unafold.rna.albany.edu/?q=mfold was used to predict Watson–Crick-based intramolecular DNA folding patterns and thermodynamics of aptamers and designated antidote sequences [20]. Duplex thermodynamics were assessed using the two-state melting (hybridization) application on the DINAMelt web server available at http://unafold.rna.albany.edu/?q=DINAMelt [21]. Default settings were used for both applications, except that the Na+ and Mg2+ concentrations were set to 150 and 1 mM, respectively. In addition, for determination of duplex thermodynamics by DINAMelt, the total strand concentration was set to 1 μM.
Oligodeoxynucleotides and chemicals
The sequence of the DNA-based aptamer (HS02-52G) was as follows: 5′-GCCTCCTAACTGAGCTGTACTCGACTTATCCCGGATGGGGCTCTTAGGAGGC-3′. The sequences of the antidote molecules (AD52, AD36, AD26, and AD22) are shown in Fig. 2A. AD52 (that represents a full-length complementary sequence of HS02-52G) was used as an antidote and as a control (non-target-binding) oligodeoxynucleotide. All oligodeoxynucleotides were synthesized (200 nmol scale) and high-performance liquid chromatography purified by Ella Biotech (Martinsried, Germany). Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (Munich, Germany).
Biolayer Interferometry analysis
Biolayer Interferometry (BLI) technology was applied to determine the binding kinetics of the aptamer, HS02-52G, to APC and the zymogen PC [22]. A BLItz system (Pall Life Sciences, Dreieich, Germany) and the Blitz 1.2 software package were used for corresponding analysis. In brief, a High-Precision Streptavidin (SAX) Biosensor (Pall Life Sciences) was loaded for 2 min with 3′-biotinylated HS02-52G (using a 500 nM solution of the aptamer). Then, for determination of association rate constants (ka [kon]), the loaded biosensor was equilibrated in binding buffer [1× Dulbecco's phosphate-buffered saline (D-PBS), containing 0.9 mM CaCl2 and 0.5 mM MgCl2, pH adjusted to 7.4, 0.1% bovine serum albumin (BSA)] for 2 min, and subsequently lowered into a drop that contained recombinant (r) APC (drotrecogin alfa, Xigris®; Eli Lilly) at the indicated concentration in binding buffer. Thereafter, for determination of dissociation rate constants (kd [koff]), the biosensor was lowered into a tube containing 500 μL of binding buffer. All measurements were performed at a shaking speed of 2,200 rpm. Between analysis of increasing APC concentrations, the HS02-52G-loaded biosensor was regenerated by incubation (3 × 30 s) in 200 μL of fresh 10 mM glycine solution (pH 1.7), followed by incubation in 200 μL of binding buffer for 2 min.
Binding of HS02-52G to PC was assessed in the same way using Ceprotin® (Baxalta, Munich, Germany) as a source of PC. The concentration of PC in the used batch of Ceprotin had been previously determined by measurement of both PC activity (Berichrom Protein C Kit; Siemens Healthcare Diagnostics, Marburg, Germany) and PC antigen (Vidas Protein C Kit; Biomerieux, Nürtingen, Germany) levels in parallel with a characterized PC preparation [Haematologic Technologies, Inc. (HTI), Essex Junction, VT], whereat one unit of Ceprotin was found to correspond to 3.58 μg (57.7 pmol) PC.
Activated factor VIII (FVIIIa) inactivation assay
To determine the influence of HS02-52G on APC-mediated inactivation of FVIIIa, a tenase assay was used. In brief, FVIIIa was prepared by incubation of 1 U human FVIII (Haemate P; CSL Behring, Marburg, Germany) with 0.025 U human α-thrombin (HTI) in PBS buffer (pH 7.4, 1 mg/mL BSA) in a total volume of 100 μL at room temperature. After 2 min of incubation, a final concentration of 100 μM argatroban (Mitsubishi Tanabe Pharma GmbH, Düsseldorf, Germany) was added to terminate any thrombin activity. Activated FVIII at a concentration of 0.16 U/mL was then incubated with 10 nM rAPC (Xigris) in assay buffer [20 mM Tris-HCl (pH 7.6), 137 mM NaCl, 10 μg/mL phospholipids, 5 mM CaCl2, 1 mg/mL BSA] in the absence or presence of the indicated concentrations of HS02-52G or AD52. After incubation for 30 min, 25 μL of the mixtures was transferred to the wells of black F16 FluoroNunc modules [Thermo Fisher Scientific (Nunc)] containing 3 nM human FIXa (HTI) and 333 μM Boc-Ile-Glu-Gly-Arg-AMC (Bachem, Bubendorf, Switzerland) in a total volume of 75 μL of assay buffer. Subsequently, 50 μL 25 nM human FX (HTI) in assay buffer was added to the wells and the kinetics of FXa-mediated substrate hydrolysis monitored using a Synergy 2 microplate reader (Biotek, Bad Friedrichshall, Germany). To assess the HS02-52G-neutralizing activity of the antidotes, increasing concentrations of AD molecules were introduced at the indicated concentrations after addition of HS02-52G (100 nM) to the test system.
General procedures during functional testing of aptamer and antidotes in plasma or whole blood
Citrated normal pooled plasma (NPP, in-house preparation) or whole blood taken from healthy volunteers (whole blood and plasma were obtained from healthy blood donors who gave written informed consent) was spiked with the intended molecules at the indicated concentrations and introduced to the below described basic or modified (APC-dependent) coagulation assays. For assessment of the performance of the antidote molecules, NPP or whole blood was spiked with HS02-52G and the mixture aliquoted. Subsequently, the antidote molecules were added at the indicated concentrations and the aliquots incubated at 37°C for 5 min before analysis.
Prothrombin time and activated partial thromboplastin time measurements
Plasma prothrombin time (PT) and activated partial thromboplastin time (aPTT) were measured using a fully automated coagulation analyzer (BCS XP System; Siemens Healthcare Diagnostics). For determination of the PT, 100 μL of TF reagent (Innovin®; Siemens Healthcare Diagnostics) was added to 100 μL of plasma sample and time to clot formation measured optically and given in seconds (s). For determination of the aPTT, 50 μL of plasma samples was mixed with 50 μL of the contact phase activator Actin FS (Siemens Healthcare Diagnostics) and incubated for 3 min at 37°C. Subsequently, the clotting process was initiated by the addition of 50 μL of 25 mM CaCl2 solution (Siemens Healthcare Diagnostics). Results were given as clotting times (s).
Thrombin generation assay
Thrombin generation in plasma samples was monitored by calibrated automated thrombography (CAT), as described in detail elsewhere [23]. A final TF concentration of either 1 or 5 pM was applied (PPP reagents; Stago, Düsseldorf, Germany). During the CAT assay, the generation of thrombin in the samples is monitored over time through cleavage of the fluorogenic peptide substrate, Z-Gly-Gly-Arg-AMC (Bachem). Fluorescence (λex = 390 nm/λem = 460 nm) was measured every 20 s using a Fluoroskan Ascent FL microplate reader (Thermo Fisher Scientific). The parameters derived from the resulting thrombin generation curves include the lag time (min), the peak thrombin level (nM), the time to peak (min), and the endogenous thrombin potential (ETP [nM × min]) that is represented by the area under the curve.
Whole blood clotting assay
Whole blood clotting times were measured using aPTT reagents and the semiautomated 10-channel ball coagulometer KC10 [Amelung, Lemgo, Germany (now: Diasys, Flacht, Germany)]. In brief, 100 μL of citrated whole blood and 50 μL of Actin FS were added to the system-specific cuvettes and incubated at 37°C for 3 min. Subsequently, 50 μL of 25 mM CaCl2 solution was added to start the clotting reaction. Time to detectable clot formation was measured mechanically and given in seconds.
Assessment of antidote molecules
The effectiveness of the different designed antidote molecules to reverse the APC inhibitory activity of HS02-52G was assessed on the BCS XP system using an aPTT-based assay. Plasma was spiked with HS02-52G and antidotes at the given concentrations and analyzed as described above. Instead of the starting CaCl2 solution, however, a solution of 25 mM CaCl2 in HEPES buffer (140 mM HEPES, pH 7.0, 0.1% BSA) containing 300 ng/mL (5.5 nM) rAPC (CaCl2/APC solution) was added. While the present rAPC prolonged the clotting times, this effect could be neutralized by addition of the aptamer, HS02-52G, to the samples. Accordingly, at a fixed plasma concentration of HS02-52G, the concentration-dependent neutralizing effect of the antidotes could be assessed by increasing clotting times.
Assessment of HS02-52G-based inhibition of APC and the neutralizing effect of AD22
HS02-52G and AD22 were added to NPP that had been previously spiked with rabbit TM (Sekisui, Pfungstadt, Germany) at a final concentration of 2 U/mL to achieve thrombin-dependent APC formation. Thrombin generation in the samples was initiated by TF added at a final concentration of 5 pM and monitored by CAT as described above. The generated APC cleaved FVa and FVIIIa, leading to overall reduced thrombin generation.
To assess the functionality of HS02-52G and AD22 in the whole blood matrix, citrated whole blood was spiked with HS02-52G and AD22 at the indicated concentrations and introduced to the whole blood clotting assay. Again, the CaCl2/APC solution was used as the starting reagent, leading to HS02-52/AD22-dependent clotting times.
Data analysis and presentation
Data analysis and presentation were done using Microsoft Excel and Powerpoint software (Microsoft Office 2010). Half-maximal inhibitory concentrations (IC50 values) were determined by nonlinear curve fit using the four-parameter logistic function, y = D + (A − D)/[1 + (x/C)B], where C represents the inflection point (IC50), B the hill slope, and A and D the minimum and maximum asymptote of the curve, respectively. The best-fitting values of the four parameters were determined by using the Microsoft Excel Solver add-in for iterative least squares fitting of residuals as described elsewhere [24].
Results and Discussion
During previous characterization of the APC-targeting aptamer, HS02-52G, its binding affinity was determined by filter retention analysis under equilibrium conditions [19]. In the present study, the BLI technology was applied to reveal the pharmacologically more relevant kinetics of binding of HS02-52G to and dissociation from APC [25]. As shown in Fig. 1A, the interaction of APC with immobilized HS02-52G aptamers could be detected over a wide range of APC concentrations. Using a one-to-one binding model, global analysis of gathered association and dissociation data revealed on and off rates of 1.118 ± 0.013 × 105 M−1 s−1 (kon) and 1.234 ± 0.033 × 10−3 s−1 (koff), respectively. Accordingly, using this particular test system, the derived KD (koff/kon) of HS02-52G for APC was determined as 11.0 ± 0.32 nM. In contrast, even when introduced at a concentration of 500 nM, only negligible binding of HS02-52G to zymogenic PC could be detected (Fig. 1A).

Binding kinetics, inhibitory activity, and specificity of HS02-52G.
The determined kon and koff values reveal HS02-52G as a potent inhibitor of APC with a long residence time (1/koff) of 13.5 min within the formed aptamer-APC complex [25]. In combination with its high specificity and the generally short circulatory half-life of nonmodified nucleic acid-based aptamers, these features render HS02-52G as a well-controllable drug for the management of acute APC-related bleeding complications.
Indeed, as shown in Fig. 1B, with an IC50 value of 10.2 nM, HS02-52G efficiently blocked the inactivation of FVIIIa by APC in a purified test system, while AD52, which served as a non-APC-binding DNA control, showed no effect, proving an aptamer-specific effect. Owing to the high selectivity of the aptamer over structurally similar PC and other plasma serine proteases, a comparable IC50 value was found when assessing the APC-neutralizing activity of HS02-52G in an aPTT-based plasma clotting assay (Fig. 1C).
The high selectivity of HS02-52G could be further confirmed by the fact that even high concentrations of HS02-52G (1 μM) did not influence the clotting times of various plasma or whole blood-based global coagulation assays (Fig. 1D). However, while the presence of HS02-52G slightly altered the kinetics of plasma thrombin generation as measured by CAT, similar patterns were observed when applying an equally sized DNA control molecule (AD52), indicating underlying DNA- rather than aptamer-specific effects (Fig. 1E).
Being subject to renal as well as hepatic clearance, impairment of organ function in critically ill patients may result in inadvertent accumulation of aptamers in the circulation, leading to an increased risk of thrombotic complications due to ongoing inhibition of APC. Thus, to retain control in such critical situations, the availability of an antidote strategy for fast and specific inactivation of circulating aptamer molecules is of primary importance to improve the safety of an APC-inhibiting treatment approach [26].
It has been previously described that protamine as well as other positively charged polymeric molecules may serve as universal antidotes for the reversal of aptamer activity [27]. However, although protamine is a clinically approved drug used to neutralize the anticoagulant effects of heparin, its application is accompanied by a variety of cross-reactivity-based side effects that may worsen the patients’ outcome, especially under critical clinical conditions [28]. Thus, to ensure best possible target specificity of the envisaged aptamer antidote, we decided to target and, hence, neutralize the activity of HS02-52G by complementary (antisense) DNA molecules [26,29].
The design of antisense molecules capable to efficiently disrupt the functional structures of HS02-52G was determined by the stable hairpin configuration of the aptamer (Fig. 2A). On the one hand, the in silico predicted high negative value of change of Gibbs free energy (ΔG = −63.5 kcal/mol) indicated tight association between HS02-52G and its full-length complementary counterpart, AD52. On the other hand, however, the stable intramolecular folding patterns of both HS02-52G (ΔG = −6.8 kcal/mol) and, accordingly, AD52 (ΔG = −6.6 kcal/mol) may compromise efficient duplex formation (Fig. 2A).
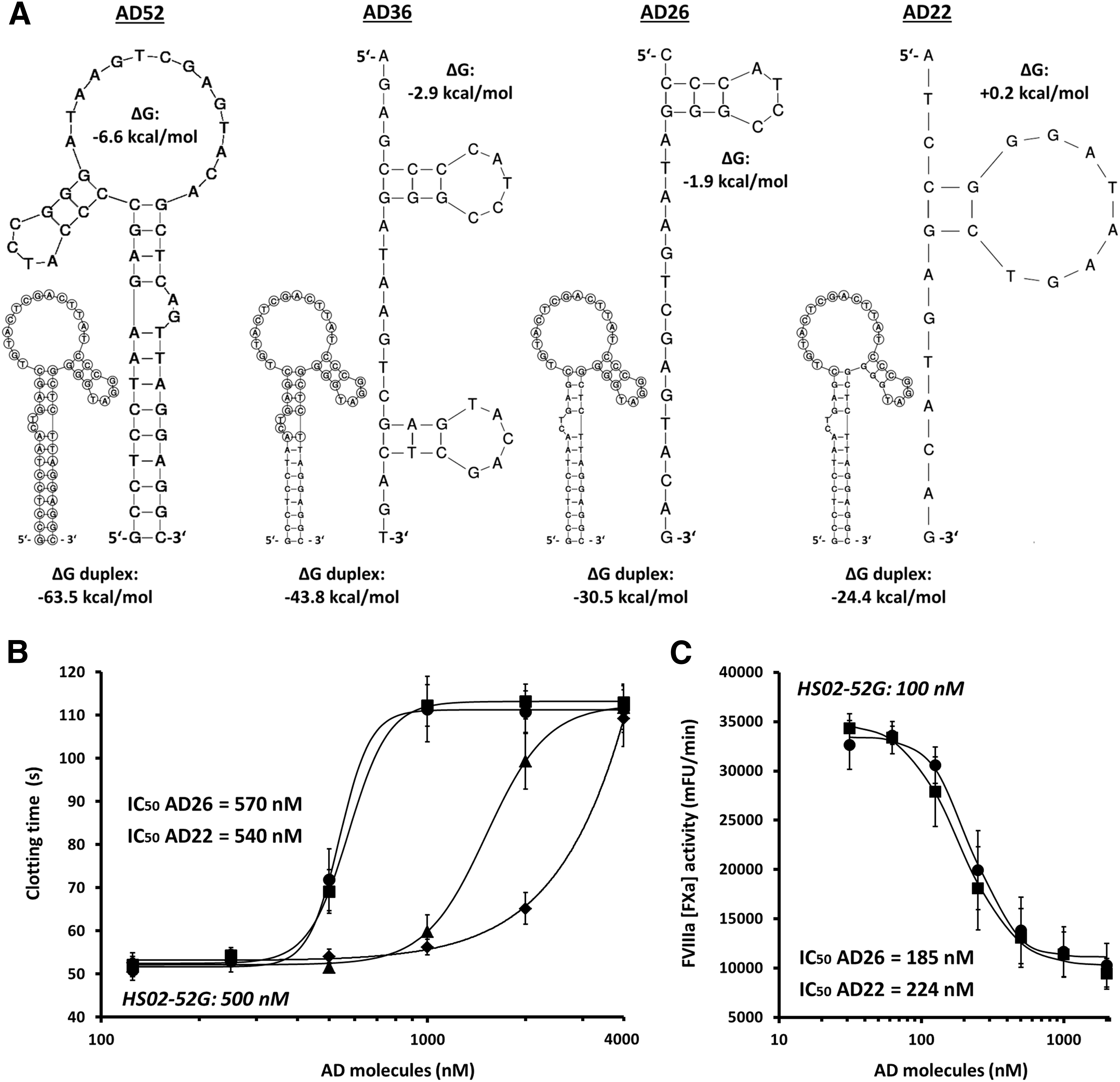
Design of HS02-52G antisense molecules (AD series) for neutralization of HS02-52G functional activity and initial assessment.
Thus, beside AD52, we also designed a series of truncated antidote molecules that target different numbers of nucleotides (AD36, AD26, and AD22) within the extended loop structure of HS02-52G. In comparison with AD52, the predicted intramolecular folding patterns of these antidote variants are of subordinate importance, bringing along better premises for duplex formation in general (Fig. 2A). However, it remained unclear to which extent these shorter molecules have the ability to hybridize to their target regions embedded within folded HS02-52G aptamers.
To comprehensively investigate both the APC inhibitory activity of HS02-52G and the efficiency of the different HS02-52G antidotes, different plasma- as well as whole blood-based clotting assays were used. For initial assessment of the antidote molecules, the aPTT-based plasma clotting assay was applied. In brief, HS02-52G was used at a saturating plasma concentration of 500 nM to completely neutralize APC-prolonged clotting times (cf. Fig. 1C). As shown in Fig. 2B, increasing concentrations of the different AD variants were applied to this test system to assess the HS02-52G-neutralizing capacity of these molecules, as reflected by increasing clotting times. Up to a concentration of 1 μM, no significant effects could be observed for the full-length antisense molecule, AD52, or the truncated version, AD36. In contrast, the more truncated antidotes, AD26 and AD22, yielded potent HS02-52G neutralization patterns, whereat comparable IC50 values were obtained (IC50 = 570 and 540 nM, respectively; Fig. 2B). Similar results that confirmed the comparable performance of AD26 and AD22 were found by testing both molecules with respect to neutralization of the APC inhibitory effect of 100 nM HS02-52G in the buffer-based FVIIIa inactivation assay (cf. Fig. 1B). When introducing AD26 or AD22 at increasing concentrations, IC50 values for neutralization of HS02-52G activity were found to be 185 and 224 nM, respectively (Fig. 2C).
For evaluation of antidote-controlled inhibition of APC under more physiological conditions, HS02-52G and AD22 were introduced to a plasma-based thrombin generation assay, namely CAT [23]. With an IC50 value of 31.9 nM, HS02-52G potently restored thrombin generation in NPP that was spiked with TM to give rise to the endogenous generation of anticoagulant APC during the run of the assay (Fig. 3A). As shown in Fig. 3B, in accordance with initial assessments, AD22 efficiently (IC50 = 165 nM) neutralized the APC inhibitory effect of HS02-52G that was applied at a concentration of 100 nM.

Further evaluation of the performance of HS02-52G and AD22 in plasma and in whole blood. Upper panels: CAT assay.
Further characterization of the identified aptamer–antidote pair involved the application of a modified aPTT-based whole blood assay. In this study, HS02-52G effectively normalized APC-prolonged whole blood clotting times (IC50 = 7.5 nM; Fig. 3C). In addition, neutralization of 100 nM HS02-52G by AD22 could be readily achieved (IC50 = 217 nM; Fig. 3D).
The main advantages of HS02-52G over the previously described small-molecule-based APC inhibitors include its high target specificity as well as the availability of an antidote strategy in case of unwanted accumulation in the circulation under pathophysiological conditions. If a long-lasting inhibitory effect as would be necessary, for example, for the adjuvant treatment of hemophilia, is desired, modification of HS02-52G by, for example, pegylation, to increase the circulatory half-life of the aptamer would be necessary for application of the aptamer for such clinical purposes [29,30]. Due to the chemical synthesis of aptamers, however, modifications, for example, needed for further conjugation, can be readily incorporated [31].
In summary, in the present article, HS02-52G has been introduced as a highly specific APC inhibitor with a fast onset of action and a predicted long residence time. It could be demonstrated that the aptamer retains its high inhibitory activity in plasma and whole blood and that its functional activity within these matrices can be effectively reversed by the short antisense molecule, AD22. In the first place, these features qualify the novel native aptamer–antidote pair as a save candidate treatment option for acute APC-related bleeding complications as, for instance, potentially observed in TIC [8].
Footnotes
Acknowledgment
The authors thank Simone Gasper for expert technical assistance.
Author Disclosure Statement
No competing financial interests exist.