
Abstract
The pathophysiology of sickle cell disease (SCD) is dependent on the polymerization of deoxygenated sickle hemoglobin (HbS), leading to erythrocyte deformation (sickling) and vaso-occlusion within the microvasculature. Following deoxygenation, there is a delay time before polymerization is initiated, during which nucleation of HbS monomers occurs. An agent with the ability to extend this delay time or slow polymerization would therefore hold a therapeutic, possibly curative, potential. We used the Systematic Evolution of Ligands by Exponential Enrichment (SELEX) method to screen for HbS-binding RNA aptamers modified with nuclease-resistant 2′-fluoropyrimidines. Polymerization assays were employed to identify aptamers with polymerization-inhibitory properties. Two noncompeting aptamers, DE3A and OX3B, were found to bind hemoglobin, significantly increase the delay time, and reduce the rate of polymerization of HbS. These modifiable, nuclease-resistant aptamers are potential new therapeutic agents for SCD.
Introduction
S
Current therapies shown to alter the severity of the disease such as bone marrow transplantation and blood transfusion are hindered by complications and limitations [8,9]. Hydroxyurea, a drug that ameliorates symptoms of SCD by increasing the production of fetal hemoglobin (HbF), which inhibits polymerization of HbS [10], decreases vaso-occlusive complications such as painful crises and acute chest syndrome, but can be associated with adverse side effects and is not an effective therapy for all patients [11]. Accordingly, alternative therapies are needed for SCD.
In this study, we have developed two aptamers as potential therapeutic agents and models for future drug development. Aptamers are small, single-stranded nucleic acid molecules that fold into unique structures, allowing them to bind to molecular targets with high specificity and affinity. They are advantageous, as they are reliably synthesized and easily stored, generally nonimmunogenic and nontoxic, can be antidote-controlled, and are modifiable to improve their therapeutic qualities [12–14]. Additionally, an aptamer's small size can enable it to bind to a specific site on a protein, altering the function of that site, without affecting other functions of the protein. For example, Blake et al. [15] have developed aptamers that bind specifically to plasminogen activator inhibitor-1 (PAI-1), blocking its interaction with vitronectin, without affecting its antiproteolytic activity. In this study, we demonstrate that two noncompeting RNA aptamers, DE3A and OX3B, bind to HbS, increasing the delay time and slowing the rate of polymerization of deoxygenated HbS (deoxyHbS) in a hypoxic cell-free system.
Materials and Methods
Preparation of hemoglobin
Heparinized venous blood from discarded blood samples of untransfused SCD patients and human blood for HbF experiments were acquired with approval by the Institutional Review Board of the Johns Hopkins Hospital School of Medicine. Erythrocytes from SCD patients (HbSS or HbS-β0 thalassemia) were washed five times with phosphate-buffered saline, hemolyzed in 3.5 volumes of distilled water, and stromata were removed by centrifugation at 20,000 g for 25 min. Hemoglobin-rich extract was dialyzed into 0.05 M Tris-HCl, pH 8.3, and purified HbS was obtained by separation on a DEAE Sephadex A-50 anion exchange column, developing with a gradient of 0.05 M Tris-HCl, pH 8.3 to 0.05 M Tris-HCl, pH 7.3. The appropriate peak was collected and shown by high-performance liquid chromotography to contain 95% HbS.
The collected fractions were dialyzed against 2 mM HEPES, pH 7.4 for the Systematic Evolution of Ligands by Exponential Enrichment (SELEX) process, and against 1 M potassium phosphate buffer, pH 7.1 for use in the polymerization assays, and stored at −80°C. For HbF, hemoglobin-rich extract was acquired as above from a patient with 99.5% HbF, and dialyzed in H2O. Hemoglobin concentrations were measured by Drabkin's method [16]. Proportions of hemoglobin at different oxidation states were determined by the method of Benesch et al. [17] corrected for an extinction coefficient of 11.0 [18].
Selection of aptamers through SELEX
The initial RNA oligonucleotide library comprised the sequence 5′-GGGAGGACGAUGCGG(N40)CAGACGACUCGCUGAGGAUCCGAGA-3′, where N40 represents a random sequence of 40 nt. The RNA incorporated modified nuclease-resistant nucleotides 2′-fluorine-dCTP and 2′-fluorine-dUTP. From this starting RNA library, four initial rounds of selection were performed, in which a partially deoxygenated solution of HbS was the target (Fig. 1). To deoxygenate the HbS before incubation, the preparation was thawed, exposed to a vacuum by injection into a vacuum tube with a septum cap, and rocked at room temperature for 1 h. The hemoglobin was then removed from the tube, and incubated with RNA immediately at 37°C for 5 min at a ratio of 3 moles RNA per mole of protein in round 1, increasing to 5 moles RNA per mole of protein by round 4.

Schematic of SELEX process to select for oxyHbS- and deoxyHbS-binding aptamers. The initial RNA library was subjected to four rounds of positive selection in which the aptamer pool was incubated with deoxyHbS, followed by recovery of bound aptamers and reverse transcription to generate cDNA. The cDNA was amplified and transcribed to create an aptamer pool for the following round. OxyHbS was the target in round 5 and both the bound and unbound fractions were recovered in this round. The bound pool underwent 9 more rounds (6–14) of positive selection against oxyHbS. The unbound pool underwent 10 subsequent rounds to select for deoxyHbS-targeting aptamers. Because of the difficulty in maintaining high levels of deoxyHbS in the binding steps, rounds of positive selection (6, 8, 10, 12, 14) against a deoxyHbS preparation were alternated with rounds of counter selection (7, 9, 11, 13, 15) against an oxyHbS preparation, with the unbound aptamers from the counter selection rounds collected and advanced for further selection. cDNA, complementary DNA; deoxyHbS, deoxygenated sickle hemoglobin; dsDNA, double-stranded DNA; oxyHbS, oxygenated sickle hemoglobin; SELEX, Systematic Evolution of Ligands by Exponential Enrichment.
Bound RNA was collected by capturing the RNA/protein complexes on a nitrocellulose membrane, eluting, and extracting the RNA. Reverse transcription was performed on the eluted RNA, followed by polymerase chain reaction. Transcription was performed and the resulting aptamer pool was used in the subsequent selection round. To prevent nonspecific binding of the aptamers to nitrocellulose, the aptamer pool was incubated with nitrocellulose alone before the binding reaction in each round. Preliminary tests showed that the vacuum-deoxygenated lysates contained approximately 18%–28% deoxyHbS, which decreased during subsequent incubation. The remainder was a mixture of oxygenated HbS (oxyHbS) and methemoglobin S (metHbS); therefore, the first four rounds enriched for aptamers targeting both the T- and R-states. By round 4, binding to oxyHbS was 28%.
Following round 4, the screening strategy was modified to capture two pools of aptamers: those that bound oxyHbS and those that bound deoxyHbS. Therefore, in round 5, oxyHbS was the target. Freshly thawed HbS, in which measurements of the oxidation states showed 81%–90% to be oxyHbS, with variable amounts of deoxyHbS and metHbS, was used in the binding reaction. Incubation was carried out at room temperature for 10 min at a ratio of 5 moles RNA per mole of protein. Following binding, the aptamer/HbS complexes were captured on nitrocellulose, and the unbound aptamers were also collected and recovered by butanol extraction. At this juncture, the two collected pools (bound and unbound fractions) proceeded in two separate selections (Fig. 1).
The bound pool was used to enrich for aptamers that bind to oxyHbS. For this selection, oxyHbS was the target in rounds 6–14, and binding was carried out at room temperature for 10 min at a ratio of 5 moles RNA per mole of protein in round 6, increasing to 9 moles RNA per mole of protein by round 14. Only bound aptamers were recovered for further selection during these rounds; unbound aptamers were discarded (Fig. 1). By round 9, binding of this pool to oxyHbS was 57%.
The unbound pool from round 5 was used in the second selection, designed to enrich for aptamers that bind to deoxyHbS. To remove those aptamers that bind to the oxyHbS still present following deoxygenation, a counter selection was applied: positive selection against deoxyHbS, as described for rounds 1–4, was alternated with counter selection against oxyHbS. Bound aptamers were collected for further selection after binding with deoxyHbS, and unbound aptamers were collected for further selection following binding with oxyHbS (Fig. 1). Ten rounds were completed in this manner, with binding performed at room temperature for 10 min at a ratio of 5 moles RNA per mole of protein in round 6, increasing to 7 moles RNA per mole of protein by round 15. After round 11, binding of aptamers to deoxyHbS was low, at 12%, perhaps due to the low percentage of deoxyHbS in the target solution or losses with counter selection.
Rounds 1–7 were performed in low-salt binding buffer [20 mM HEPES pH 7.4, 50 mM NaCl, 2 mM CaCl2, 0.01% bovine serum albumin (BSA)], and low-salt wash buffer (20 mM HEPES pH 7.4, 50 mM NaCl, 2 mM CaCl2). From round 8 on, selections were performed in high-salt binding buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM CaCl2, 0.01% BSA) and high-salt wash buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM CaCl2).
Cloning, sequencing, and RNA preparation
At round 11 and following the final round of selection, complementary DNA (cDNA) was removed for cloning and sequencing of individual aptamers. cDNA was amplified and cloned into the pCR2.1-TOPO vector using the TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA). Sanger sequencing was carried out at the Johns Hopkins Genetic Resources Core Facility. Aptamers containing modified nucleotides were generated by transcription using the DuraScribe T7 Kit (Epicentre, Madison, WI). RNA was recovered with the RNA Clean and Concentrator-5 Kit (Zymo Research, Irvine, CA), eluted in H2O and further concentrated by vacuum when necessary. Consensus sequences were identified using a ClustalW multiple sequence alignment [19]. Secondary structures with minimum free energy were predicted with Mfold software [20].
Polymerization assays
HbS in 1 M potassium phosphate buffer pH 7.1 was thawed on ice, concentrated in an Amicon Ultra 30K centrifugal filter tube (Millipore, Billerica, MA) at 4°C, and kept on ice. Aptamers in distilled H2O were denatured at 80°C for 1 min, allowed to cool to room temperature, and placed on ice. Distilled water alone, or an unrelated aptamer of the same length, flanking sequences, and modified nucleotides, were used as negative controls. Sodium dithionite was employed at a 4:1 dithionite:heme molar excess to ensure maximal deoxygenation of HbS. Sodium dithionite was prepared in deoxygenated potassium phosphate buffer, thereby preventing the generation of reactive oxygen byproducts [21]. To deoxygenate sodium dithionite powder, it was placed in a tube with a rubber septum cap and flushed with nitrogen gas [22]. Potassium phosphate buffer was deoxygenated similarly in a separate tube. Sodium dithionite stock solution and subsequent dilutions were then made using a Hamilton gas-tight syringe to transfer buffer from one tube to another and kept on ice [22].
All components were added on ice to a closed quartz cuvette (Starna Cells, Atascadero, CA) that had been flushed with nitrogen gas, and the temperature increased to 37°C. The final concentrations of all components in the cuvette were 0.12 mM HbS (heme), 0.48 mM sodium dithionite, and 0.012 mM aptamer in 1.45–1.55 M potassium phosphate, pH 7.42–7.80. Buffer concentration and pH were consistent within each experiment. Turbidity was measured at regular intervals with a Beckman DU-640B spectrophotometer (Beckman Coulter, Inc., Brea, CA) at a wavelength of 700 nm [22–24]. Measurements were also taken at 540, 560, and 576 nm to determine the proportions of hemoglobin derivatives present. For concentration–response assays, only the final concentration of aptamer was varied. For experiments involving HbF, HbF dialyzed in H2O was added in increasing concentrations, keeping the total volume and final concentration of HbS constant.
Electron microscopy
At the 78-min time point during the polymerization assay, where the H2O control was approaching maximal polymerization and the solutions with aptamer had just begun to polymerize, 20 μL was removed from each solution with a Hamilton syringe and added to 1 mL of deoxygenated 2% glutaraldehyde. Samples were spun at 7,000 g for 10 min and brought to 30 μL to concentrate fibers. One microliter of each sample was adsorbed to a glow-discharged carbon-coated 400-mesh copper grid (Electron Microscopy Sciences, Hatfield, PA) by flotation for 2 min. Grids were quickly blotted then rinsed in three drops (1 min each) of 50 mM Tris buffered saline. Grids were negatively stained in two consecutive drops of 0.75% uranyl formate, blotted, then quickly aspirated to get a thin layer of stain covering the sample. Grids were imaged on a Phillips CM-120 transmission electron microscope operating at 80 kV. Images were taken with an 8 megapixel CCD camera (Advanced Microscopy Techniques, Woburn, MA).
Double filter-binding assays
Double filter-binding assays were performed with the various RNA pools and individual aptamers (DE3A, OX3B, and the control aptamer). RNA was dephosphorylated with bacterial alkaline phosphatase (Invitrogen) and 5′ end-labeled with γ-32P-ATP (PerkinElmer, Waltham, MA) using T4 polynucleotide kinase (New England Biolabs, Ipswich, MA). RNA was diluted to 2,000 cpm/μL in binding buffer, heated at 80°C for 1 min, and allowed to cool to room temperature.
For the assays targeting oxyHbS, the hemoglobin was thawed and used directly as in the SELEX process. The assays targeting deoxyHbS used FluorometHbS (FmetHbS) converted to the deoxygenated conformation as the target protein. FmetHbS was prepared following the procedure of Jayaraman et al. [25] using potassium hexacyanoferrate (III) (Sigma, St. Louis, MO) at a 10% excess, with dialysis in 0.2 M sodium phosphate buffer, pH 6.8 [26], followed by the conditions described by Jayaraman et al. [25]. Sodium fluoride (Sigma) was added at a 2:1 molar excess. Final buffer exchange into 2 mM HEPES, pH 7.4 was achieved with an Amicon Ultra 30K centrifugal filter tube (Millipore).
Each protein dilution was in 15 μL of high-salt binding buffer; low-salt binding buffer was used for the initial library and round 4. The FmetHbS dilution series also contained a 15 × molar excess of inositol hexaphosphate (IHP; Sigma), an allosteric effector that stabilizes the T quaternary structure [25,27]. Five microliters of labeled RNA in binding buffer were added to each tube of the dilution series for a final concentration of 0.1–0.2 nM, incubated at 37°C for 15 min, and passed over a nitrocellulose membrane, with the unbound RNA captured on a nylon membrane. Total labeled RNA on both the nitrocellulose and nylon membranes was determined by counting radioactivity using a liquid scintillation counter (Beckman Coulter, Inc.). The percentage of labeled RNA bound to protein was calculated for each dilution, and the raw data were corrected for background binding of radiolabeled RNA to the nitrocellulose filter.
Competition binding assays
Radiolabeled aptamers were prepared as described for the double filter-binding assays (see previous section). Freshly thawed, nondeoxygenated HbS (7.5 μM) was incubated with 5 μL of labeled RNA (0.1–0.2 nM final concentration) in high-salt binding buffer at 37°C for 15 min in the presence of varying concentrations (0–20 μM) of unlabeled aptamer. To determine if DE3A and OX3B bind to the same site, we performed binding assays in the presence of increasing concentrations of unlabeled heterologous aptamer or unlabeled homologous aptamer. Following incubation, the bound complex was captured on a nitrocellulose membrane, and the unbound RNA was captured on a nylon membrane. Total labeled RNA on both the nitrocellulose and nylon membranes was determined by counting radioactivity using a liquid scintillation counter (Beckman Coulter, Inc.). The percentage of labeled RNA bound to hemoglobin was calculated for each dilution, and the raw data were corrected for background binding of radiolabeled RNA to the nitrocellulose filter. Data were normalized to the binding of labeled aptamer in the absence of cold aptamer, which was set at 100%.
Results
Aptamer pools selected using oxyHbS and deoxyHbS are predominately distinct
Our goal was to select for aptamers that, when bound to HbS, would inhibit polymerization under hypoxic conditions. To preclude any false assumptions regarding which hemoglobin structure may prove to be the best target for an effective aptamer, two separate aptamer pools were generated: one against deoxyHbS and one against oxyHbS. The selection scheme is shown in Fig. 1 and described in detail in Materials and Methods section.
Binding studies against oxyHbS showed that the initial RNA library had a total binding of 7% at a hemoglobin concentration of 10 μM, which increased to 28% by round 4, with the oxyHbS-targeting pool achieving maximal binding of 57% at round 9. In contrast, by round 11, the binding of the deoxyHbS-targeting pool to deoxyHbS was 12%. Since the target always consisted of mixed hemoglobin species, the binding results were only used as a general indication that affinity to HbS had increased since the initial library. Despite the low binding of the deoxyHbS-targeting pool to deoxyHbS, we sequenced aptamers from both pools, to determine if some of the individual aptamers in the deoxyHbS-targeting pool would be higher affinity than suggested by the pooled binding experiments.
Individual aptamers were sequenced by cloning the cDNA at round 11 and following the final round of selection. A total of 92 clones were sequenced, with 60 unique sequences represented (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/nat). Thirty-four unique aptamers were generated in the deoxyHbS-binding selection and 26 in the oxyHbS-binding selection. There were two consensus sequences specific to the deoxyHbS-targeting pool, and two specific to the oxyHbS-targeting pool, with the exception of one aptamer in each pool that contained a consensus sequence found in the other pool. Thus, the aptamers selected against deoxyHbS are predominately distinct from those selected against oxyHbS.
Identification of individual aptamers that inhibit polymerization of deoxyHbS
Individual aptamers were amplified and evaluated for their ability to inhibit the polymerization of deoxyHbS in an oxygen-depleted solution. In this system, the addition of dithionite consistently resulted in solutions of 90%–94% deoxyHbS. An idealized polymerization tracing from this type of assay is shown in Fig. 2A. The delay time, Td, reflects the time required for the formation of nuclei [22].
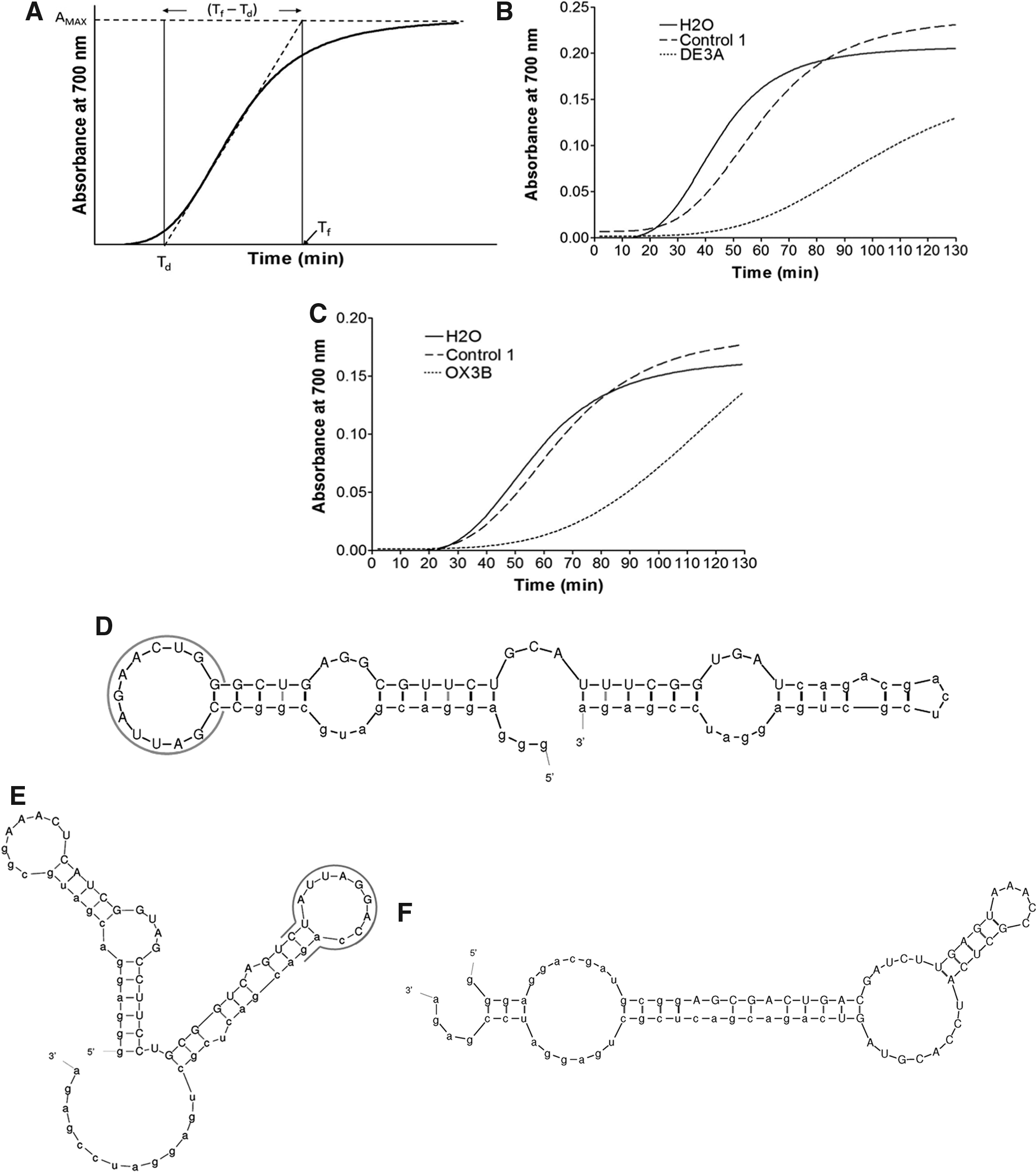
Inhibition of polymerization by aptamers DE3A and OX3B.
Two aptamers were found that consistently and significantly inhibited polymerization: DE3A, generated from the selection targeting deoxyHbS, and OX3B, generated from the selection targeting oxyHbS. Other consensus family members also inhibited polymerization, but DE3A and OX3B displayed the greatest inhibition within their families (data not shown). Of note, the short sequence AUUAG appeared within both aptamers' consensus sequences (Table 1). Delay times and slopes of polymerization in the presence of either DE3A or OX3B were compared with those obtained in the presence of a control aptamer and with no aptamer present (water control) (Fig. 2B, C). The delay time in the presence of either DE3A or OX3B was significantly longer compared with the controls. Additionally, both aptamers significantly reduced the slope of the polymerization curve during exponential growth, as compared with either the control aptamer or water alone. These results indicate that both DE3A and OX3B inhibit the kinetics of HbS polymerization by extending the time required for nucleation and slowing the rate of polymerization. Aptamer variable region sequences are shown in Table 1, and predicted secondary structures are shown in Fig. 2D–F.
Constant regions are shown in lower case; variable regions are in upper case. The DE3A and OX3B consensus sequences are underlined.
To demonstrate that our result was not simply due to protein salting out in high-concentration phosphate buffer or some other structural artifact, we employed electron microscopy to visualize the reaction products at the 78-min time point. We confirmed that HbS fibers were present in each sample, and that fibers were branching in a heterogeneous manner (Fig. 3A–C). Branching was seen in the H2O control and in the presence of both DE3A and OX3B, although not represented in Fig. 3 for DE3A. A random inspection of the entire surface of the grids established that the fibers were generally equally distributed across each grid, and that fibers were present in a greater quantity in the H2O control than in the samples containing aptamer (Fig. 3D–F). No further quantitation was done, as the process of staining for electron microscopy is known to break the fibers, affecting the results [28]. No other obvious qualitative differences in fiber structure were observed in the presence of aptamer. Our findings are also consistent with the findings of Wang et al. [29], who found that fibers formed in 1.5 M phosphate buffer have the same structures as those formed in 0.5 M phosphate buffer.
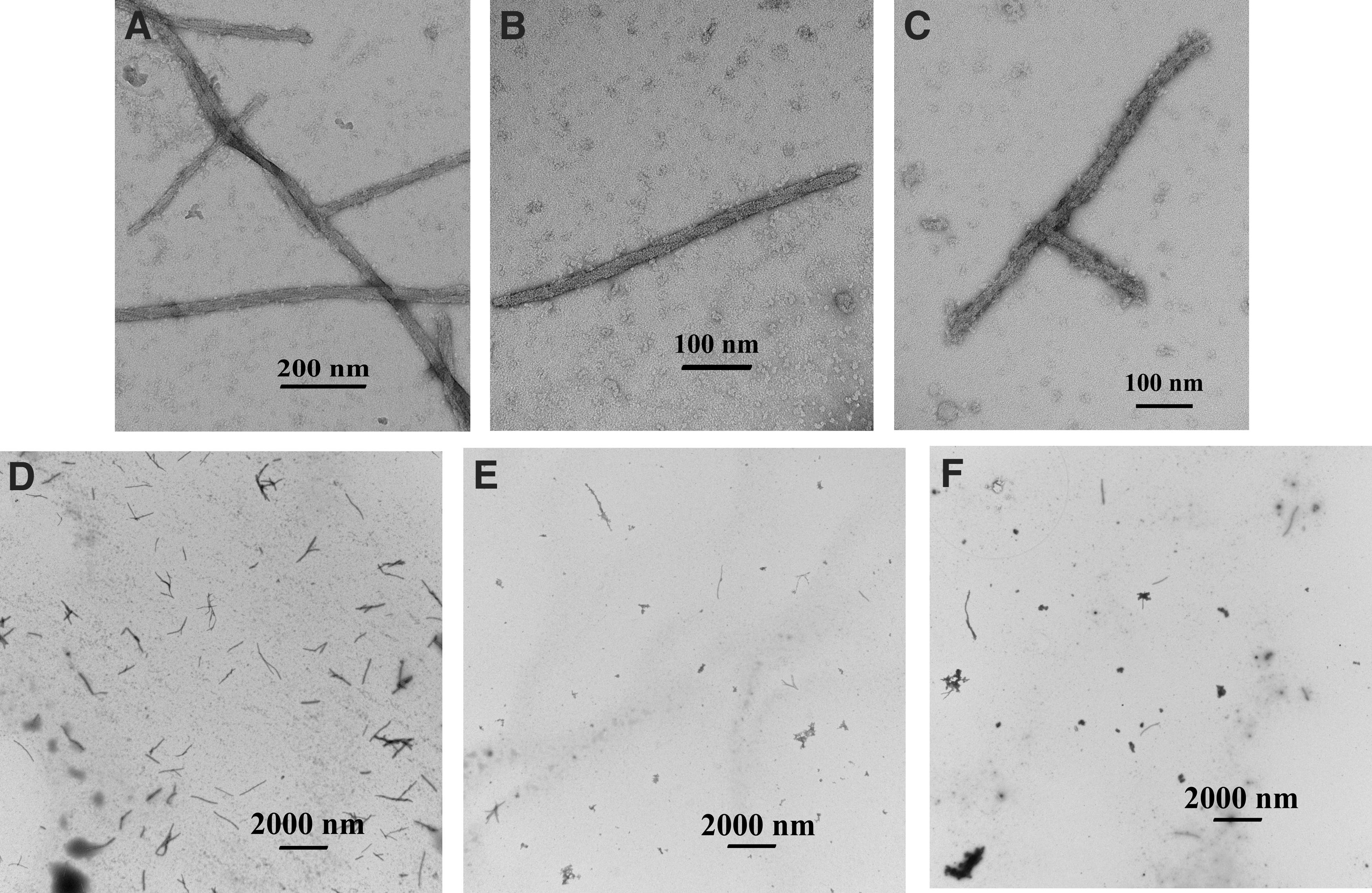
Electron micrographs showing fibers present in the polymerization assays. Fibers were fixed and stained for transmission electron microscopy at the 78-min point of the polymerization assay. The entire surface of each grid was randomly examined and representative micrographs are shown for each sample, at a magnification of
Aptamers DE3A and OX3B bind HbS
Aptamers DE3A, OX3B, and the control aptamer were assayed for their binding affinities to both oxyHbS and deoxyHbS (Fig. 4). In these assays, FmetHbS was used in place of deoxyHbS. FmetHbS is advantageous because, with the addition of IHP, the content of T-state hemoglobin is 72% or more [25] in the nonoxygen-depleted environment of the binding assay, as compared with 18%–28% for vacuum-deoxygenated HbS. DE3A showed nearly identical binding affinities to FmetHbS and oxyHbS, with respective Kd values of 1.68 and 1.74 μM. OX3B bound to oxyHbS with a Kd of 3.56 μM and to FmetHbS with a Kd of 8.57 μM. These Kd values indicate that each aptamer maintains its affinity for HbS as the protein changes conformation, although OX3B may exhibit a slightly higher affinity for oxyHbS. There was no binding between the control aptamer and hemoglobin in either conformation (Fig. 4).
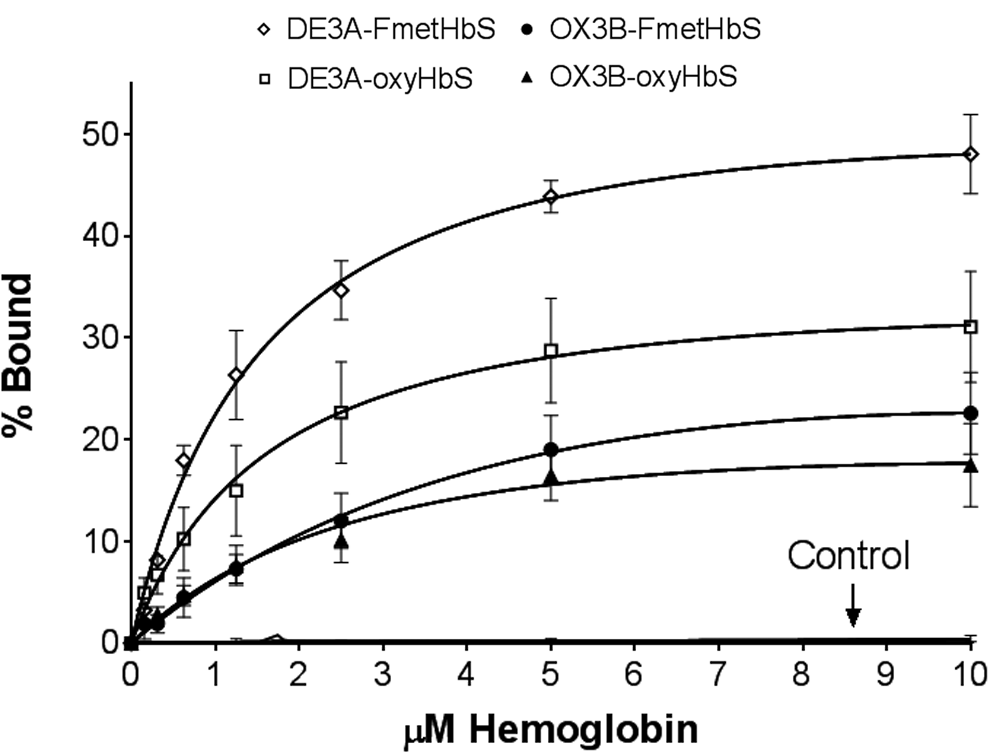
Saturation binding curves for individual aptamers. Binding of DE3A to FmetHbS, Kd = 1.68 μM; binding of DE3A to oxyHbS, Kd = 1.74 μM; binding of OX3B to FmetHbS, Kd = 8.57 μM; binding of OX3B to oxyHbS, Kd = 3.56 μM. The control aptamer exhibited no binding to either conformation. Each data point was performed in triplicate. Dissociation constants determined with GraphPad Prism 7 software (GraphPad Software, Inc., La Jolla, CA) for total saturation binding: Y = X × BMAX/(Kd + X) + NS × X. Error bars represent standard error of the mean. FmetHbS, Fluoromet sickle hemoglobin.
Concentration response of delay times and polymerization rates
The relationship between aptamer concentration and rate of polymerization is shown in Fig. 5A. At an aptamer concentration of 12 μM (an aptamer:heme ratio of 1:10), the rate of polymerization was inhibited 75.4% with DE3A and 60.8% with OX3B. Although DE3A caused a higher maximal level of inhibition than OX3B, at aptamer concentrations below ∼4 μM, OX3B appeared to be more effective than DE3A in inhibiting the polymerization rate.
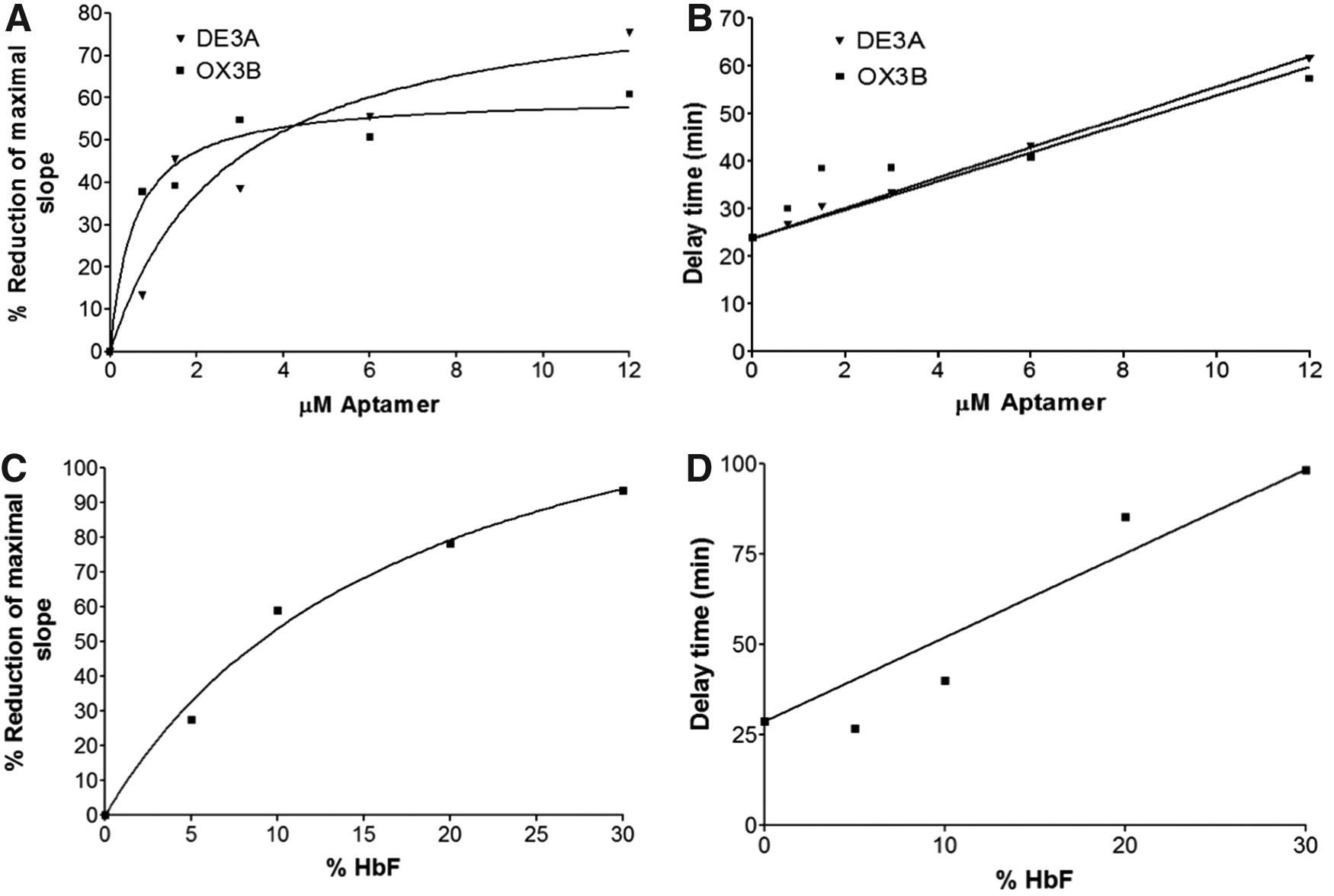
Response of delay time and polymerization rate to concentration of aptamer or HbF. HbS was combined with specified concentrations of aptamer or HbF and incubated at 37°C in the presence of dithionite under hypoxic conditions. Polymerization was measured spectrophotometrically at 700 nm. The polymerization rates, represented by the slopes in exponential growth phase, and delay times were determined from the resulting curves for each concentration of aptamer as described in Fig. 2.
The increase in delay time of polymerization with increasing aptamer concentration is shown in Fig. 5B. The delay time increased 2.6-fold with 12 μM DE3A, and 2.4-fold with 12 μM OX3B. In contrast to the aptamers' effect on polymerization rate, where nearly maximal inhibition was reached at an aptamer:heme ratio of 1:10, extrapolation of the delay time curves suggests that increasing the relative aptamer concentrations could further extend the delay time.
To determine whether this degree of inhibition is likely to have physiological significance, we compared the inhibition of polymerization by the aptamer to that of HbF in polymerization assays. With a mixture containing 10% HbF, 50% inhibition of the polymerization rate was achieved, with almost complete inhibition at 30% HbF (Fig. 5C). In comparison, ∼50% inhibition of the polymerization rate was seen with either aptamer at a concentration of 4 μM; at three times this concentration, ∼60% and 75% inhibition was seen with OX3B and DE3A, respectively. The delay time with 12 μM aptamer was ∼2.5 times greater than the delay time without aptamer. A similar extension of the delay time was also seen with the addition of approximately 15%–20% HbF (Fig. 5D). This suggests that the aptamers could inhibit the polymerization of HbS at the same order of magnitude as HbF, but higher concentrations and ratios of aptamer may be necessary for maximal effect.
Aptamers DE3A and OX3B do not compete
To determine if DE3A and OX3B bind to different sites or the same site on HbS, competition binding assays were performed in which labeled aptamer was incubated with HbS in the presence of increasing amounts of unlabeled homologous or heterologous aptamer. As shown in Fig. 6A, binding of labeled DE3A decreased from a normalized value of 100% to 32% as the concentration of unlabeled DE3A increased. Similarly, binding of labeled OX3B was reduced from 100% to 20% in the presence of increasing amounts of unlabeled OX3B (Fig. 6B). In contrast, the binding of neither labeled aptamer was significantly affected at even the highest concentration of unlabeled heterologous aptamer (Fig. 6A, B). This suggests that DE3A binds a site on the HbS molecule that is different than the OX3B binding site.

Competition binding assay. Labeled DE3A or OX3B
A peak in binding is seen below 5 μM of unlabeled aptamer in both instances, where the competing aptamers are different (Fig. 6A, B). This peak was present in each assay, and not simply caused by the contribution of one anomalous result. This increase in binding could be due to a conformational change induced by the binding of the other aptamer. It is also possible that the labeled and unlabeled aptamers are interacting with each other.
Discussion
The ability to block the polymerization of HbS would, in theory, be curative, and is an attractive target for drug development. The aim of this work was to discover aptamers which, when bound to HbS, would inhibit the polymerization of HbS and thereby have the potential to reduce the sickling of HbS-containing erythrocytes. Two such aptamers, DE3A and OX3B, have been identified. Each aptamer slows homogeneous nucleation, as the delay times with an aptamer:heme ratio of 1:10 were extended 2.6-fold with DE3A and 2.4-fold with OX3B. Additionally, once polymerization was initiated, both significantly slowed the rate of bulk polymer formation.
Variation in delay time has been associated with the severity of disease [5,30,31]. Sickle cells can occlude capillary beds, where blood flow is most restricted, or in the venules, where adhesion to endothelial cells presents a further opportunity for occlusion [32,33]. Even partial inhibition of polymerization could have important therapeutic effects; the longer the delay of polymerization, the more likely it is that the erythrocyte will pass through the capillaries and venules. Polymerization delay times within erythrocytes are very sensitive to physiological conditions and thus vary from patient to patient [31,34,35], potentially leading to variability in disease manifestation among individuals with SCD. An agent with the ability to extend delay times could shift the balance toward a less severe disease course. Our results suggest that the identified aptamers could extend the delay time and further slow the rate of growing polymers enough to potentially allow a therapeutically significant proportion of cells to escape occlusion.
In this study, achieving preparations consisting entirely of native deoxyHbS for selection procedures presented challenges. Ultimately, the efficacy of individual aptamers was evaluated based on each aptamer's ability to inhibit polymerization, regardless of the origin of its selection. Each aptamer (DE3A and OX3B) binds to both oxygenated and deoxygenated hemoglobin with similar affinities. This suggests that they each bind to structures that change very little during the conformational shift between R-state and T-state. This could be a beneficial quality in vivo, as the aptamer, once bound, would remain bound, rather than undergoing detachment at each conformational shift.
Our data suggest that aptamers DE3A and OX3B each bind to a different site on the HbS molecule. It is not yet known where those binding sites are located. The aptamers share the short nucleotide sequence AUUAG within their respective consensus sequences (Table 1). In both cases the short sequence is part of a loop in the secondary structure (Fig. 2D–E), suggesting that the aptamers' binding sites may share some similarity. Future structural studies will elucidate the binding sites. It could be advantageous to have two polymerization-inhibiting aptamers that bind different sites on the same molecule, as this may confer the ability to form a bivalent aptamer with the potential for considerably increased affinity and improved function. Ahmad et al. [36] found that the affinity of a bivalent aptamer with an optimized linker region increased ∼200-fold over the individual parental aptamers, improving its functional ability. Soule et al. [37] demonstrated that a bivalent aptamer was more effective at inhibiting coagulation than either parental aptamer alone.
Our experiments suggest that, as an estimate, half-maximal inhibition of the rate of polymerization of HbS by aptamer would produce an effect roughly equivalent to 10% HbF, greater than the average increase in HbF (3.6%), or final level of HbF (8.6%) achieved with hydroxyurea in The Multicenter Study of Hydroxyurea in Sickle Cell Anemia trial [38]. Higher concentrations could potentially cause greater effects. This presupposes that delivery of the aptamers into circulating erythrocytes could be achieved, a hurdle which will need to be addressed. Given the high molar ratios required to see effects on polymerization of HbS, our current aptamers would most likely need to be concentrated in erythrocytes to be useful in vivo; given that the aptamers bind to hemoglobin, this may be possible.
We attempted to transfect DE3A and OX3B aptamers into sickle erythrocytes using Lipofectamine 3000 (Invitrogen) to examine their effect on sickling under hypoxic conditions, with variable results (data not shown). Previous studies have suggested that cells of hematopoietic origin are difficult to transfect [39]; it is possible that we did not consistently transfect aptamers into erythrocytes at a concentration high enough to inhibit sickling. In future studies, we will investigate methods to facilitate internalization of these aptamers using methods that can also be applied in vivo, such as linking them to small molecules that are internalized by erythrocytes without affecting the integrity of the cell membrane.
An important benefit of employing aptamers as therapeutic agents is that they are easily modified. Our aptamers incorporate 2′-fluoro nucleotides to protect against nuclease degradation [14]. Macugen, an aptamer that has been approved by the U.S. Food and Drug Administration for the treatment of age-related macular degeneration, is an example of an effective, similarly modified aptamer [40,41]. Further modification of the nucleotide sequence or creation of a multivalent aptamer could improve binding affinities and potentially increase the ability to inhibit polymerization [36,37,42]. Alternatively, it may be possible to truncate our existing aptamers while retaining inhibitory function, which might increase cellular uptake.
At present, we can only speculate on the mechanism by which aptamers inhibit polymerization, but given that the aptamers may bind different sites, and DE3A appears to affect both the rate and extent of HbS polymerization, whereas OX3B seems to inhibit only the rate of polymerization, DE3A may act by binding HbS monomers, whereas OX3B may simply inhibit polymerization of the elongating filaments. Neither aptamer appeared to induce nucleation, which might be a property of a species that could bind to the growing end of filaments and induce the filamentous conformation in monomers, as occurs with actin and its filamentous end-binding proteins [43]. Further experimentation will be required to determine where the aptamers bind to hemoglobin and how they may affect hemoglobin function.
In summary, we have identified two aptamers with the ability to inhibit the polymerization of HbS in lysates under hypoxic conditions. With the ability to deliver these aptamers intracellularly to erythrocytes at high concentrations, these two HbS polymerization-inhibiting RNA aptamers could potentially reduce or eliminate the consequences of sickling in patients with SCD. Further investigations to find an effective delivery mechanism in vivo and to introduce modifications that can improve potency could increase the potential of these aptamers as new candidate therapeutic agents in treating SCD; alternatively, further investigation of the structure and binding sites for these molecules could lead to the development of small molecules with similar effects, which could be used as therapeutic agents.
Footnotes
Acknowledgments
The authors are grateful to James Tonascia (University of Pennsylvania) for assistance in cloning, to Michael Delannoy (Johns Hopkins University School of Medicine Microscope Facility) for assisting with electron microscope analyses, and to Sue Dixon (Johns Hopkins University School of Medicine) for coordinating blood sample acquisition.
Author Disclosure Statement
All authors have filed a patent on the aptamers discovered in this work and the use of aptamers to inhibit the polymerization of HbS, as well as other potential uses of the aptamers described in this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.