
Abstract
RNA has enormous potential as a therapeutic, yet, the successful application depends on efficient delivery strategies. In this study, we demonstrate that a designed artificial viral coat protein, which self-assembles with DNA to form rod-shaped virus-like particles (VLPs), also encapsulates and protects mRNA encoding enhanced green fluorescent protein (EGFP) and luciferase, and yields cellular expression of these proteins. The artificial viral coat protein consists of an oligolysine (K12) for binding to the oligonucleotide, a silk protein-like midblock S10 = (GAGAGAGQ)10 that self-assembles into stiff rods, and a long hydrophilic random coil block C that shields the nucleic acid cargo from its environment. With mRNA, the C-S10-K12 protein coassembles to form rod-shaped VLPs each encapsulating about one to five mRNA molecules. Inside the rod-shaped VLPs, the mRNAs are protected against degradation by RNAses, and VLPs also maintain their shape following incubation with serum. Despite the lack of cationic surface charge, the mRNA VLPs transfect cells with both EGFP and luciferase, although with a much lower efficiency than obtained by a lipoplex transfection reagent. The VLPs have a negligible toxicity and minimal hemolytic activity. Our results demonstrate that VLPs yield efficient packaging and shielding of mRNA and create the basis for implementation of additional virus-like functionalities to improve transfection and cell specificity, such as targeting functionalities.
Introduction
T
Because of the transient nature of mRNA-dependent protein expression, for use as a therapeutic outside the area of vaccination, repeated administrations will be required, which mandates the use of nonviral vectors that do not elicit immunity. Nonviral transfection agents that have been investigated for plasmid DNA (pDNA), in particular (cationic) polymers and cationic lipids, can also be used for nonviral transfer of small interfering RNA (siRNA) and mRNA. Generally, lipid-based formulations have been found to work better than polymer-based formulations [1–3]. Indeed, for siRNA delivery, the formulation of lipid-based nanoparticles with high transfection efficiency and an adequate safety profile has developed into a highly mature technology also for in vivo applications [5]. However, even for the lipid-based formulations, rapid clearance from the blood and poor biodistribution remain crucial problems that need to be solved before use in targeted delivery to tumors or specific organs [6]. Therefore, there is a continued need for new approaches that will offer the tunability required to improve the in-vivo efficacy of RNA therapeutics.
Recently, we have developed a family of de novo designed artificial viral coat proteins [7]. When mixed with DNA (linear dsDNA, supercoiled pDNA, and ssDNA), these proteins coassemble to form rod-shaped virus-like particles (VLPs). Each VLP encapsulates a single nucleic acid molecule and protects it against enzymatic degradation. The main aim of that work was to show that the essential features of the assembly of rod-shaped viruses can be mimicked in terms of three functional blocks, each having a simple repetitive amino acid sequence: one for DNA binding, one for protein–protein interactions, and one for colloidal stabilization. VLPs encapsulating pDNA had an appreciable transfection efficiency for HeLa cells, and low toxicity
The DNA binding block in the design of the artificial viral coat proteins is a simple oligolysine (K12) that should not be restricted to complexation of DNA. Therefore, we intended to explore whether these proteins will also form VLPs with mRNA, and thereby form a versatile scaffold for the development of novel mRNA transfer agents. With this in mind, we, in this study, investigated the VLP formation of artificial viral coat proteins with mRNA, including an assessment of the stability of the encapsulated mRNA against enzymatic degradation, and of their transfection efficiency for HeLa and HEK293 cells, in vitro.
Materials and Methods
Materials
The protein C-S10-K12 was produced and purified as described before [7]. The enhanced green fluorescent protein (EGFP) mRNA (996 nucleotides, 1 mg/mL in 10 mM Tris-HCl, and pH 7.5) was purchased from TriLink Biotechnologies (L-6301, Tebu-Bio, Heerhugowaard, the Netherlands). For the electrophoretic mobility shift assay (EMSA), we used EGFP mRNA 5 meC Ψ (5-methylcytidine, pseudouridine), while for the other experiments, the mRNA was capped (Cap 0) and polyadenylated, mimicking fully processed mature mRNA. Luciferase NanoLuc mRNA, derived from the deep sea shrimp Oplophorus gracilirostris, was synthesized from a DNA template. Cassettes with codon-optimized genes expressing yeast Nluc (yNluc) were assembled in the context of the dominant drug resistance marker kanamycin. The Nano-Glo luciferase assay with Nano-Glo substrate and assay buffer was obtained from Promega (Leiden, the Netherlands). Agarose, 6 × DNA loading dye, and RNase A/T1 mix (2 mg/mL of bovine pancreas RNase A and 5,000 U/mL of Escherichia coli RNase T1) were purchased from Thermo Fisher Scientific (Breda, the Netherlands). SYBR gold DNA gel stain (10,000 × concentrate in DMSO) and TrackIt 1 kb Plus DNA ladder were purchased from Life Technologies. Lipofectamine Messenger Max was obtained from Thermo Fisher Scientific. RPMI 1640 medium with Glutamax I, Dulbecco's modified Eagle's medium plus Glutamax I, with or without phenol red, Opti-MEM medium, and HEPES-buffered saline (HBS) were acquired from Gibco, Invitrogen, Thermo Fischer Scientific. Fetal calf serum (FCS) was obtained from Gibco or Euro Clone (Uden, The Netherlands). Trypsin/EDTA was obtained from PAN Biotech (Aidenbach, Germany). Red blood cells (RBCs) were obtained from RBC units of blood group 0, Rhesus-positive donors that had been collected and processed according to standard Dutch blood bank protocols, including leukoreduction and storage in saline–adenine–glucose–mannitol. HeLa cells and HEK293 cells were obtained from the DSMZ (Braunschweig, Germany). All other chemicals (analytical grade) were obtained from Sigma-Aldrich (Zwijndrecht, the Netherlands). Flat-bottom 96-well, black/clear, sterile tissue culture plates, used for nanoluciferase assay, were obtained from BD Bioscience (Breda, the Netherlands). Cover slides with eight wells for confocal microscopy were obtained from Ibidi (Martinsried, Germany). Centrifuge filters with molecular weight cutoffs of 3 kDa, 300 kDa, and 1 MDa were obtained from VWR International, Amsterdam, the Netherlands.
Preparation of mRNA VLPs
Fresh stock solutions of 1 mg/mL of the C-S10-K12 protein were prepared by dissolving a weighted amount of the protein powder in 10 mM phosphate buffer containing 0.1 mM dithiothreitol (DTT), filtered using 3 kDa cutoff centrifuge filters (VLP buffer). The C-S10-K12 has a unique cysteine at its C-terminus, and DTT was added to prevent the formation of disulfide bridges. Aliquots of the dissolved protein stock were stored at −20°C for later use. The mRNA stock solutions were diluted 20 times in the VLP buffer to a 20 μL solution containing 100 ng/μL of EGFP mRNA or NanoLuc mRNA. Next, VLP and the respective mRNAs were mixed at N/P ratio's of 3, 6, or 9, where N/P is defined as the ratio of moles from the primary ammonium ion (N) of the lysine chains and the phosphate groups (P) in the mRNA. The mRNA VLP solution was mixed by vortexing for ∼1 min and heated for 1 h at 65°C with occasional vortexing to ensure full dissolution of the protein into monomeric subunits capable of assembling into VLPs. To form mature VLPs, the complexes were incubated for 24 h at room temperature. Finally, VLP nanoparticles containing 1 ng/μL of EGFP mRNA in 500 μL or 5 ng/μL NanoLuc mRNA in a total volume of 100 μL were obtained to be used in the transfection experiments.
Positive control Lipofectamine-mRNA complexes
Lipofectamine Messenger Max was used according to the manufacturer's protocol. First, 0.3 μL of Lipofectamine Messenger Max was diluted in 5 μL of Opti-MEM and incubated for 10 min. Separately, 1 ng/μL of NanoLuc mRNA or EGFP mRNA was diluted 50 times in the Opti-MEM medium. Finally, equal volumes of the mRNA and Lipofectamine solutions were mixed to form the complexes at a final mRNA concentration of 10 ng/μL of mRNA.
Atomic force microscopy imaging
As a substrate for atomic force microscopy (AFM) imaging of dried complexes, ultraflat silicon wafers (Siltronic, Munich, Germany) were used. Wafers were cut into 1 × 1 cm squares and cleaned by sonication in a 48% (v/v) ethanol/MilliQ mixture for 30 min–1 h. Before use, silicon substrates were dried with nitrogen and plasma-cleaned. For AFM imaging, 5 μL of sample was applied onto freshly cleaned silicon substrates and left to dry for ∼3 min. Substrates were carefully rinsed with 1 mL of filtered MilliQ water to remove salts and nonadsorbed particles and slowly dried with nitrogen. Samples were analyzed using a Digital Instruments Nanoscope V equipped with a non-conductive silicon nitride probe with a spring constant of 0.24 N/m in the ScanAsyst imaging mode. Images were recorded at 0.977 Hz and at 512–1,024 samples/lines. Images were processed using NanoScope Analysis 1.20 software. A second-order flattening was applied for all images. Lengths of particles were analyzed with ImageJ software. Concentration of mRNA used for the AFM experiments was 1 μg/mL. For the experiments where VLPs were postincubated with phosphate-buffered saline (PBS), RPMI-1640 tissue culture medium and serum (10% FCS), and 150 μL of VLP solution (1 ng/μL, N/P = 3) were mixed with 150 μL postincubation solution and incubated for 2 h before AFM imaging.
Gel electrophoresis assays
For EMSA, samples were prepared by mixing protein stock solutions previously heated at 70°C for 10 min, with the EGFP mRNA in TAE 1× buffer and incubated at room temperature for 1 or 24 h. Approximately 50 ng of mRNA (per lane) was loaded in 1% agarose gels, at different protein to DNA ratios. Samples were run at 95 V for 60 min. Next, gels were incubated for 10–20 min with SYBR gold according to the instructions provided by the manufacturer, and the gels were imaged. For assessing the stability of mRNA-VLPs against RNAses, a stock solution of mRNA VLPs (mRNA concentration 1 ng/μL, N/P = 3) was prepared as described above. Subsequently, samples of 9 μL of mRNA-VLP stock solution were incubated with 1 μL RNAse A/T1 mixture for the indicated periods of time at room temperature. In a separate experiment, it was established that under these conditions, the degradation of bare RNA was complete in about 1 min. After incubation, 2 μL of 6 × loading buffer was added to the samples. Samples were mixed by repeated pipetting up and down, and were electrophoresed in 1% agarose gels at 100 V for 1.5 h. RNA bands were visualized with SYBR Safe stain. Gels were imaged with Bio-Rad Gel Doc EX Imager.
Dynamic light scattering
Dynamic light scattering was performed using a Zetasizer Nano-ZS with a HeNe laser (4 mW, 633 nm) at a scattering angle of 173°. Scattering was performed in a small volume quartz cell and the temperature was maintained at 20°C. Sample volumes were typically 20 μL. The measuring mode was set to automatic, such that the Malvern DTS software (version 7.11) chose the optimal run numbers, and run times comprising a single sample measurement. The reported distribution of the contribution to the light scattering versus the effective hydrodynamic radius DH was calculated from the intensity autocorrelation functions by Malvern DTS software.
Cell culture
HeLa cells were grown in the RPMI 1640 medium with Glutamax I (RPMI with glutamine, sodium pyruvate, pyridoxine, and 4.5 g/L glucose) supplemented with 10% FCS. Cells plated in a T-75 culture flask and incubated in a humidified incubator with 7.5% CO2 at 37°C, were passaged every 2 to 3 days at 80% confluency. HEK 293T cells were grown in Dulbecco's modified Eagle's medium plus Glutamax I and 10% FCS. Cells were passaged after 8/10 of the culture medium was aspirated, followed by washes with 5 mL HBS, followed by incubation with 0.05% trypsin in EDTA during 5 min. Cells were spun down for 3 min at an rcf of 150 g, after which the supernatant was removed, and the cell pellet was resuspended in 10 mL of full culture medium. From the cell suspension, 1/3 of the total volume was collected, diluted four times, and plated in a new T-75 flask. All the cells used in this experiment were at passage number 11 or 12.
Nanoluciferase assays
For the NanoLuc Luciferase Assay, HeLa and HEK cells were seeded in a flat-bottom 96-well plate 24 h before the incubation with the nanoparticles at 20,000 cells/well. Before transfection/incubation with nanoparticles, cells were washed twice and maintained in 135 μL of cell culture medium with 10% serum, but without phenol red. Next, 15 μL of bare mRNA, mRNA-VLPs, or mRNA MessengerMAX complex was added to the culture medium and carefully mixed. Hence, cells were incubated for 4 h with 150 μL of cell culture medium containing 10% of NanoLuc mRNA-VLP nanoparticle solution, with a total concentration of 75 ng of mRNA. After the incubation, cells were washed with HBS and protein expression was observed after 24 and 48 h with the NanoLuc luciferase assay.
The NanoLuc luciferase assay was conducted according to the manufacturer's protocol, except that only 20 μL of the cell culture supernatant was collected and diluted five times in MilliQ. NanoLuc is secreted into the tissue culture supernatant; therefore no cell lysis is required. Afterward, the NanoGlo Luciferase Assay Reagent was prepared and an equal volume was added to the cell supernatants. The samples were distributed in a 96-well flat-bottom black/clear tissue culture plate. The bioluminescence assay readout can bring some bias if the collected samples are placed in adjacent wells. In this experiment, we placed the MessengerMax sample (positive control), at least at three wells distance from other samples. The raw luminescence units (RLU) were obtained using a Synergy 2 Multi-Mode reader and data analysis was performed with Gen5 software (BioTek). Each read was done with the luminescence detection mode, endpoint read type. The emission filter was selected for hole; the optics position was on top, and the sensitivity was 135. The luminescence for each time point t was calculated according to the manufacturer's protocol, and expressed as follows:
in which the luminescence expressed in RLU is calculated by the software. The samples were collected after 24 and 48 h of incubation. The dilution factor is the factor by which the cell supernatants are diluted for subsequent analysis.
Confocal laser scanning microscopy of EGFP expression
For the confocal laser scanning microscopy assay of EGFP expression, one day before the experiment 40,000 HeLa cells and 50,000 HEK cells were seeded in eight-well cover slides. Cells were maintained in 150 μL of cell culture medium with 10% FCS, but without phenol red. Immediately before transfection, the cells were washed and then maintained in 150 μL of cell culture medium without phenol red, and with or without 10% FCS, as indicated. Next, 150 μL of bare EGFP mRNA, mRNA-VLPs, or mRNA MessengerMAX complex was added. Hence, mRNA concentration in contact with the cells was 2 × diluted compared to the stocks. Cells were incubated with 150 ng of mRNA or mRNA complexes for 4 h before washing with HBS, and then kept in culture for either 24 or 48 h with the cell culture medium with 10% FCS and 20 mM HEPES. Next, confocal microscopy was performed using a Leica SP5 microscope with an HCX PL APO 63 × N.A. 1.2 water immersion lens (Leica, Mannheim, Germany). During image acquisition, the cells were maintained in the same 20 mM HEPES-containing full cell culture medium, without phenol red, at 37°C, in which they had been cultivated. EGFP was excited using an argon ion laser at 488 nm and emission was collected between 500 and 550 nm.
For a semiquantitative comparison of EGFP fluorescence intensity of different samples, with roughly similar cell density, images were obtained at the same settings of the microscope, and the average intensities of background-corrected cell-associated fluorescence was determined using Fiji [8].
Cytotoxicity of mRNA complexes toward HeLa cells
The acute toxicity of complexes of mRNA with C-S10-K12 toward HeLa cells was quantified using a resazurin assay [9]. HeLa cells were cultured as described above. Cells were transferred and seeded in a 96-well plate at a density of 8,000 cells/per well, one day before performing the cytotoxicity assay. Each well contained the cells in 50 μL RPMI-1640 supplemented with 2 mM L-glutamine and 10% FCS. Stock solutions of mRNA complexes were prepared as described above. Small volumes of concentrated stock solutions of EGFP mRNA complexes at N/P = 3, were added to the HeLa cells in the 96-well plate. Volumes of mRNA complexes added to the cells yielded final mRNA concentrations between 0 and 25 ng/μL. Cells were incubated with the mRNA complexes for 3.5 h. Next, the mRNA-containing medium was removed and replaced with 200 μL resazurin (0.1 g/L in RPMI-1640 supplemented with 2 mM L-glutamine and 10% FCS). Cells were incubated with resazurin for an additional 2 h. Subsequently, fluorescence intensity measurements (excitation at 540/25, emission at 620/40 nm) were performed on a BioTek Synergy 2 plate reader. Nontreated cells and RPMI-1640 supplemented with 2 mM L-glutamine and 10% FCS served as a control and blank, respectively. Cell viability was calculated using the following formula:
where Isample, Icontrol, and Iblank are the fluorescence intensities of the sample, the control, and the blank, respectively.
Hemolysis induced by mRNA VLPs
Five hundred microliters of erythrocyte suspension was centrifuged in an Eppendorf tube for 3 min at 3,000 RPM and the supernatant was removed. Next, RBCs were resuspended in 500 μL Ringer solution (125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 32 mM HEPES, 5 mM glucose, and 1 mM CaCl2) and centrifuged for 1 min at 4,000 RPM. The supernatant was removed and this procedure was repeated again. The pellet was resuspended in the remaining supernatant to obtain the final suspension of washed RBCs. 2 × 107 RBCs were transferred to a new Eppendorf tube and were supplemented with Ringer solution to a final volume of 150 μL. EGFP mRNA-VLPs at N/P = 3 were added to the RBCs at concentrations corresponding to concentrations of 0–25 ng/μL of mRNA. Cells were incubated with the mRNA VLPs for 2 h. Samples were regularly shaken to prevent precipitation. Cells incubated with 10 mM phosphate buffer were used as negative control. RBCs lysed by incubation with MilliQ were used as positive control. After 2 h of incubation, RBCs were centrifuged for 3 min at 3,000 RPM and the supernatant was collected. Next, 60 μL of the supernatant was transferred to a 96-well plate and MiliQ water was added to a final volume of 300 μL. The absorbance A of each sample was measured at 405 nm and the degree of hemolysis was calculated with the following formula:
Results
VLP formation
The artificial viral coat protein, abbreviated as C-S10-K12, has been described before [7]. Its architecture is illustrated in Fig. 1. In brief, the triblock amino acid-polymer features a C-terminal oligolysine (K12) for binding to nucleic acids. The silk-like midblock S10 = (GAGAGAGQ)10 can stack into a rigid filament, which forms the backbone of the rod-shaped VLPs. Finally, colloidal stability and interactions of the VLPs with the external environment are determined by an N-terminal 400 amino acid, hydrophilic random coil polypeptide “C” with a high proportion of glycines, prolines, and other uncharged hydrophilic residues [10].
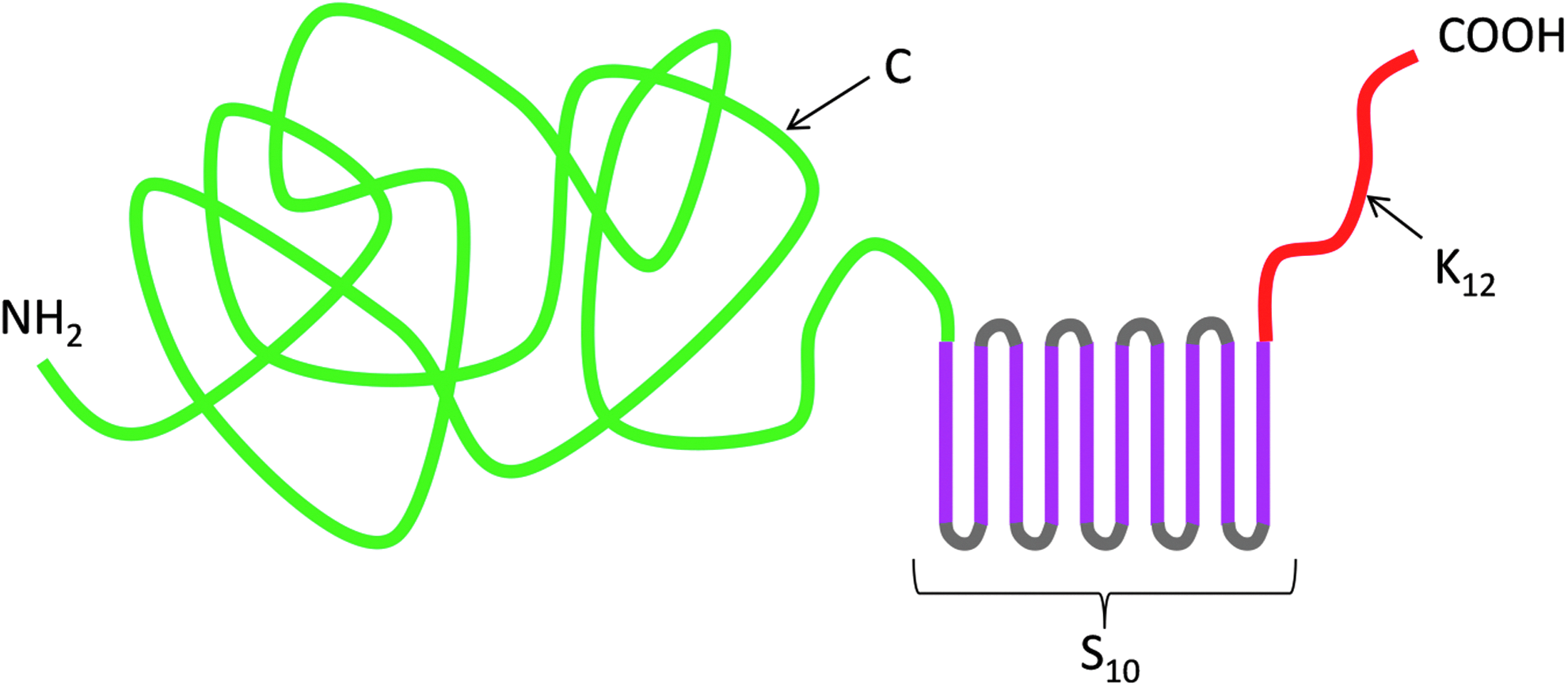
Architecture of the C-S10-K12 artificial viral coat protein. In green is the N-terminal “C” domain, a 400 amino acid, hydrophilic random coil polypeptide. In pink is the silk-like midblock S10, a 10-fold tandem repeat of the octapeptide S = GAGAGAGQ. Finally, in red is the C-terminal dodecalysine (K12) nucleic acid binding domain.
In our previous work on the formation of VLPs with double-stranded DNA, we have established that the kinetics of VLP formation is a strong function of both the absolute concentration of DNA template and the DNA: protein charge stoichiometry. The charge stoichiometry is specified in terms of the ratio N/P where N is the number of nitrogens on the side chains of the lysine residues of the K12 binding block, and P is the number of phosphate groups of the nucleic acid template. For DNA concentrations of around 1 μg/mL, a stoichiometry of at least N/P = 3 appears to be necessary for fully mature VLPs to grow in about 10 h.
To assess to which degree these observations can be transferred to messenger RNA, an EMSA at various N/P ratios was performed with a 996 nt EGFP mRNA (Fig. 2). Complexation was assessed after 24 h. Full complexation was achieved at N/P ≥ 3. We conclude that, like for the case of DNA, N/P > 3 appears to be necessary to coat all the mRNA with the artificial capsid protein.

Binding of C-S10-K12 artificial viral coat protein to mRNA. Electrophoretic mobility shift assay with 996nt EFGP mRNA at different protein to mRNA ratios and different incubation times. Lane 1 is a DNA molecular weight marker, lanes 2–11 are for mRNA to protein charge ratios (N/P) of, respectively, N/P = 0, 0.2, 0.5, 0.75, 1, 2, 3, 5, 8, and 10. The amount of mRNA was 50 ng/well, solution conditions, 10 mM phosphate buffer at pH 7.4, 0.1 mM DTT. Incubation time was 24 h at room temperature. DTT, dithiothreitol.
Next, AFM was used to image complexes formed after 24-h complexation at N/P = 3. This corresponds to a protein concentration of 34.9 μg/mL (or 0.78 μM). As shown in Fig. 3a, we find rod-shaped VLPs with an average length of Lc ≈ 150 nm. From the work on VLPs with linear DNA [7], we know that to a good approximation, the stoichiometry of the VLPs is such that all nucleic acid charges are neutralized by the K12 binding blocks of the proteins and the linear density of the proteins is about 1 protein per 0.5 nm of VLP contour length. For the case of DNA, we found that each VLP encapsulated exactly one DNA molecule. If this were also the case for the 996 nt-long mRNA, we would expect VLPs consisting of about 80 proteins and contour lengths of around 40 nm. Hence, the mRNA-VLPs must contain, on average, around three mRNA molecules. The large spread can then be explained by assuming that there is a distribution of the number of mRNAs encapsulated per VLP, ranging from about one to five.

Stability of fully assembled VLPs in buffer, medium, and serum as judged from AFM imaging. VLPs were assembled at 1 μg/mL mRNA and N/P = 3 in 10 mM phosphate buffer, pH 7.4, for 24 h at room temperature. VLPs after assembly
Stability in serum is a critical parameter for the assessment of nonviral gene delivery strategies. We therefore also tested the stability of assembled VLPs in PBS, tissue culture medium, and serum-containing medium (Fig. 3b–d). The only apparent difference between the images is that for the case of serum incubation, we observed a significant absorption of serum proteins to the silicon substrates and a lower density of adsorbed VLPs. The latter may very well be caused by a competition for the adsorption to the clean silica by the abundant serum proteins and the VLPs. A quantitative analysis of the contour lengths Lc of adsorbed VLPs (Fig. 4) shows that, while there is a large spread in the VLP lengths in all cases, there appears to be no significant change in the average lengths of the VLPs due to the various incubations.
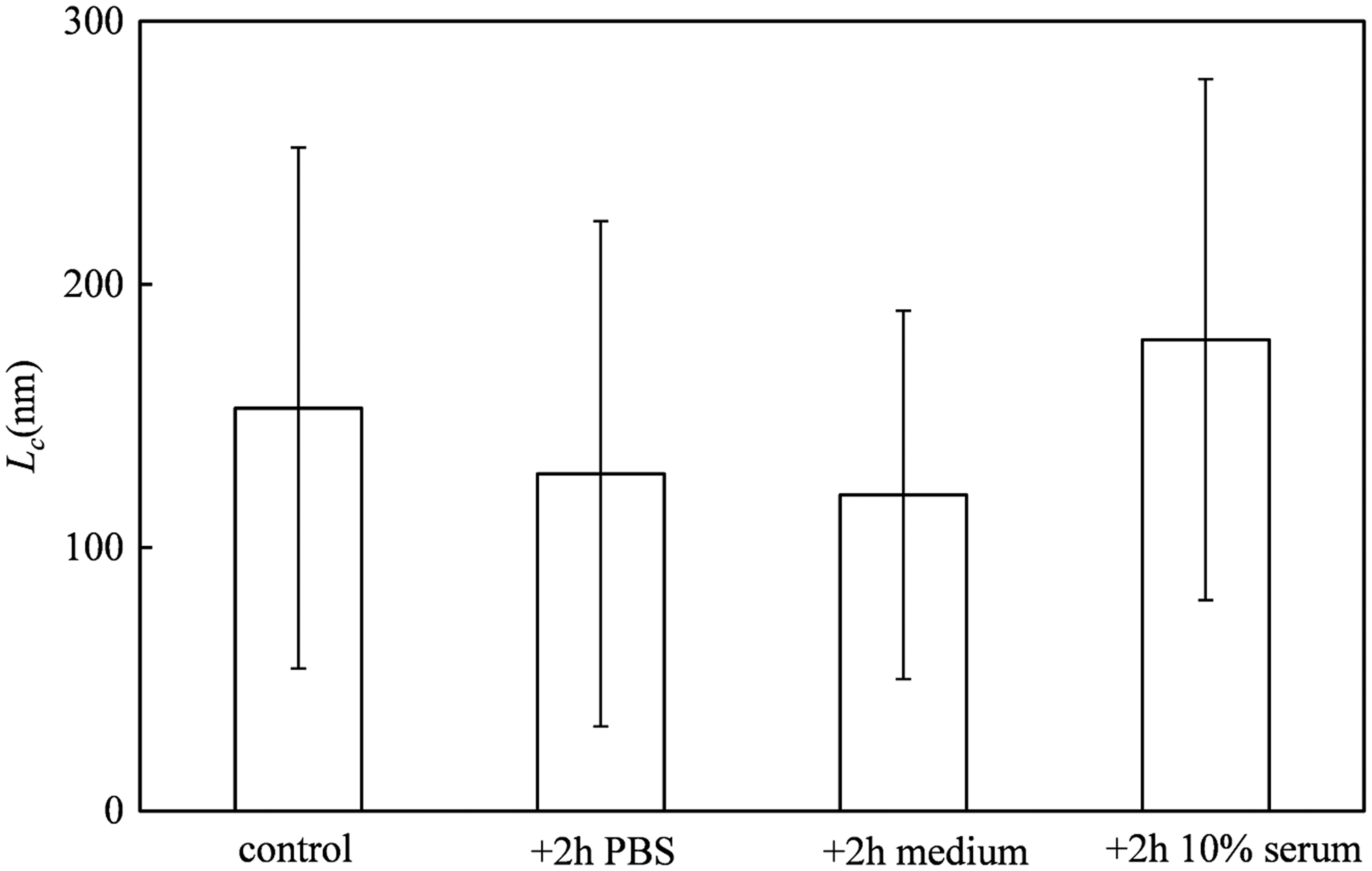
Stability of VLPs after assembly as judged from contour lengths Lc determined using AFM. Control: VLPs as formed during 24 in 10 mM phosphate buffer, pH 7.4. +2 h PBS: complexes additionally incubated with PBS for 2 h. +2 h medium: complexes additionally incubated with cell culture medium RPMI-1640 for 2 h. +2 h 10% serum: complexes additionally incubated with 10% FCS. FCS, fetal calf serum.
For a more quantitative assessment of the stability of the VLPs in serum, we turned to dynamic light scattering. To be able to observe sufficient scattering from the VLPs against a background of a concentrated solution of serum proteins, it is imperative that scattering from the serum reveals absolutely no aggregates in the same size range as the size of the VLPs. It was found that this required extensive filtering of the 10% FCS solution, first with a 1 MDa centrifuge filter to remove the largest aggregates, followed by filtering with a 300 kDa centrifuge filter to also remove smaller aggregates. Dynamic light scattering of 10% FCS filtered in this way showed a single peak corresponding to a hydrodynamic radius DH ≈ 10 nm (Fig. 5).
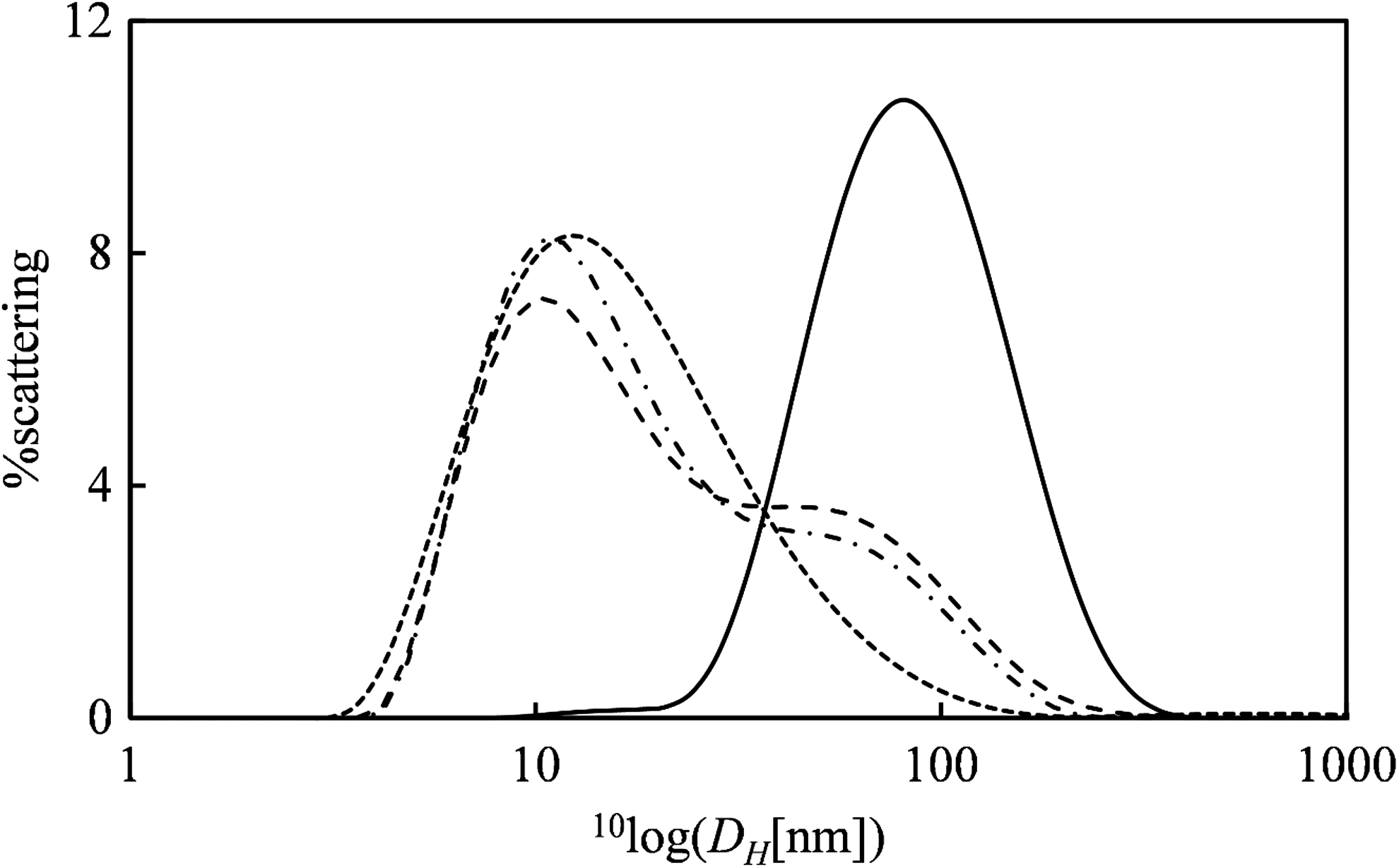
Stability of VLPs after assembly in serum as judged by Dynamic Light Scattering. Scattering intensity (%) as a function of the hydrodynamic radius DH in nm. Solid line: 7 × concentrated EGFP mRNA VLPs assembled at an mRNA concentration of 1 ng/μL. Short dashes: 10% FCS filtered using, subsequently, 1,000 and 300 kDa centrifuge filters. Long dashes: equal volume of VLPs + filtered FCS, immediately after mixing. Dash-dotted line: equal volume of VLPs + filtered FCS 2 h after mixing.
For nanoluc mRNA-VLPs at N/P = 6, at an mRNA concentration of 1 ng/μL, a single peak at the expected size of DH ≈100 nm was observed. Since it became apparent that at this concentration scattering of the VLPs would be too weak to be distinguished against the background of serum proteins, the VLPs were concentrated 7 × using a 3 kDa centrifuge filter. Following 2 × dilution with filtered 10% FCS, dynamic light scattering experiments were performed, both immediately after mixing and after 2 h of incubation of the mixed sample (at 20°C). The intensity distribution for the mixed sample had two peaks at the expected hydrodynamic diameters corresponding to the serum proteins and the VLPs (Fig. 5). Also, there was hardly any change in the scattering distributions when incubating the VLPs for up to 2 h in the presence of the serum proteins. Therefore, like the AFM experiment, the dynamic light scattering experiment demonstrates that the VLPs have appreciable physical stability in the presence of serum proteins and supports the idea that VLP binding to the silicon surface was reduced due to inhibition by serum proteins and not due to decomplexation.
Protection against RNAse
For RNA therapeutics, sensitivity toward hydrolytic breakdown is even more a concern than for DNA oligonucleotides. Therefore, we assessed the stability against degradation by RNAses using a gel retention assay. VLPs were incubated with an RNAse A/T1 mix, at a concentration that was sufficient to completely degrade the equivalent amount of free mRNA in less than 1 min. The VLPs were electrophoresed after various times of incubation with the RNAse mix of up to 24 h. The intensity of the VLP band remained the same, demonstrating excellent protection of the encapsulated mRNA (Fig. 6).
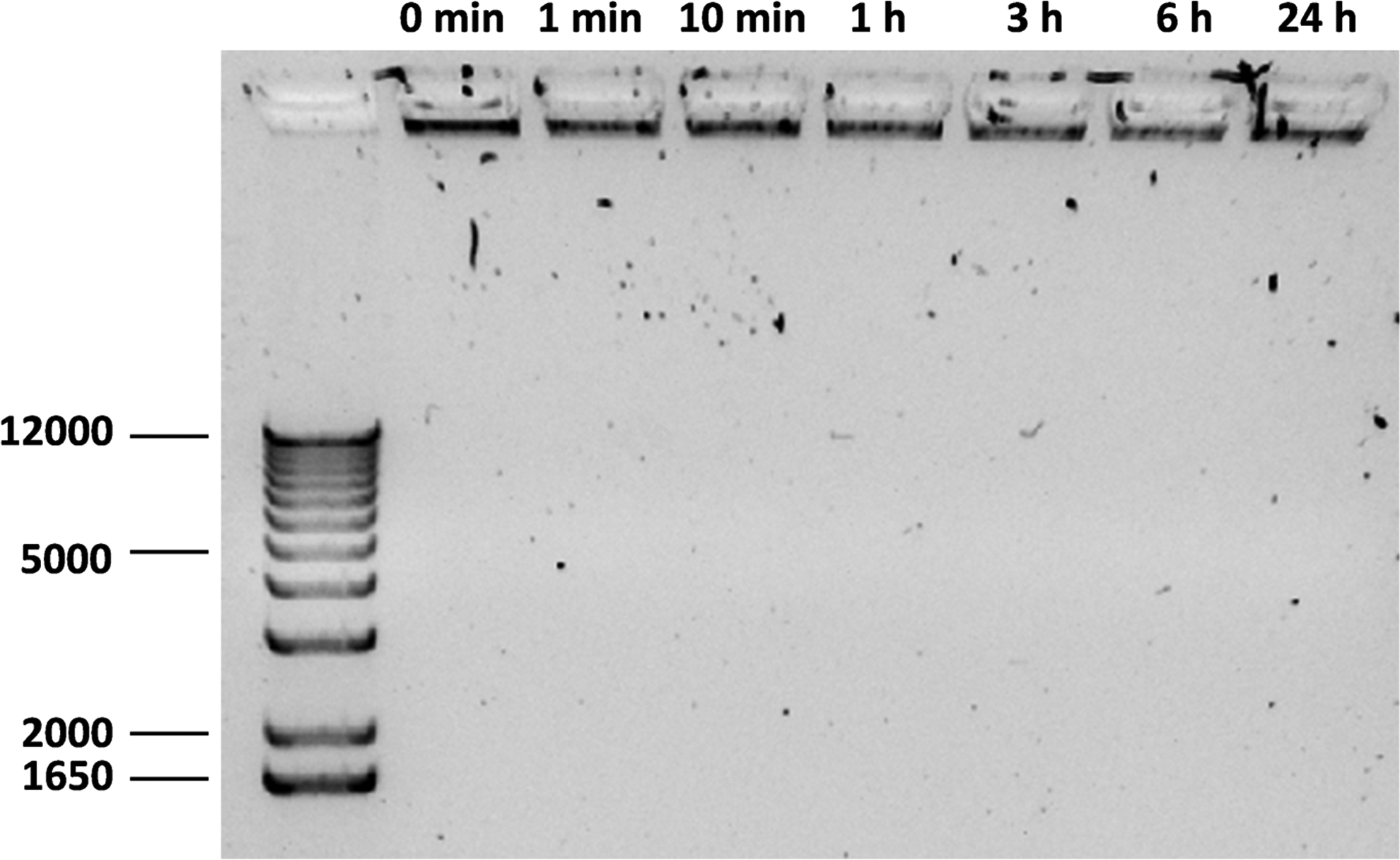
Stability of VLPs against RNAse 1% agarose gel electrophoresis of assembled mRNA VLPs after incubation with RNAse for different times. Conditions were chosen for which the equivalent amount of free mRNA was degraded in less than 1 min. The left lane is a dsDNA molar mass marker, the other lanes are VLPs encapsulating mRNA, incubated with the RNAse for times as indicated above the lanes.
Transfection experiments
Transfection experiments were done for VLPs with N/P = 3, 6, and 9, for HeLa cells, and for HEK293 cells, both in the presence and in the absence of 10% FCS, using both an EGFP encoding mRNA and a luciferase-encoding mRNA.
Nanoluc mRNA-VLPs (150 μL) were incubated with both HeLa and HEK293 cells, both in the presence and absence of 10% FCS, at a VLP concentration of 0.5 ng/μL of NanoLuc mRNA corresponding to 75 ng of mRNA. For HeLa cells, luminescence did not significantly exceed that of the bare mRNA control sample, but for the HEK293 cells, luminescence was significantly larger for the VLP samples, compared to the bare mRNA control, and signals were higher after 48 h than after 24 h (Fig. 7). For N/P = 3 in the absence of serum, the luminescence signal was about 100 × higher than the one for the bare mRNA control. However, in the presence of 10% FCS, the luminescence signal was of the same order of magnitude as for bare mRNA.
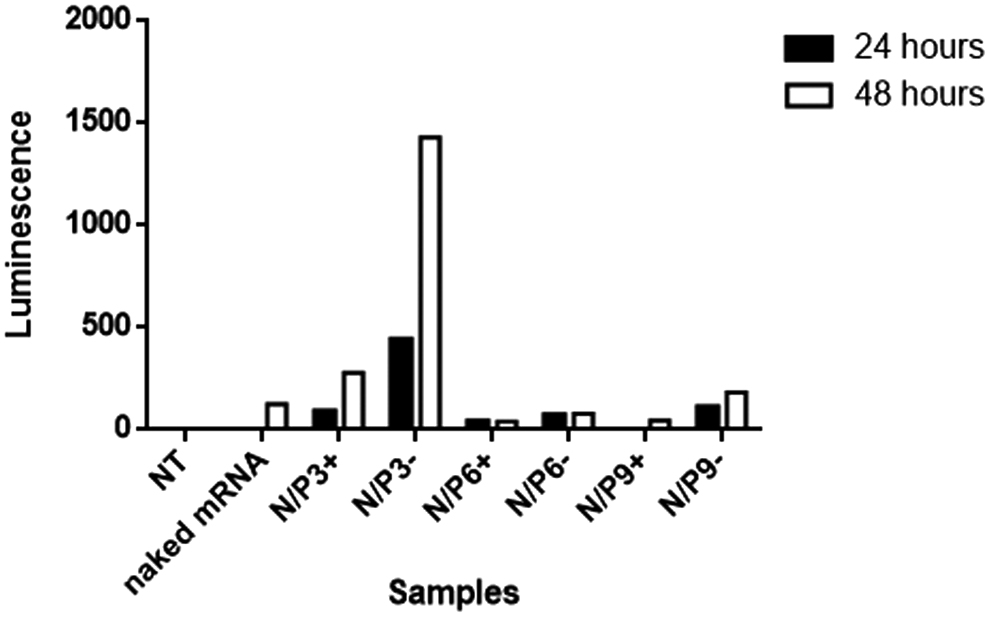
Luciferase assay for the transfection of HEK293 cells with mRNA VLPs. Luminescence of supernatants of HEK293 cells (raw luminescence units) incubated with VLPs assembled at different N/P ratios, both in the presence (+) and absence (−) of 10% FCS, at 24 and 48 h of incubation with the VLPs.
Finally, 1 ng/μL of EGFP mRNA VLP solution was diluted 2 × in the culture medium, with or without 10% FCS, on HeLa and HEK293 cells. In total, cells were incubated with 150 ng of EGFP mRNA for 4 h, followed by washing steps and analysis through confocal microscopy. Consistent with the results for the luciferase assay, for HeLa cells, there was no difference in the fluorescence of cells treated with bare mRNA or VLPs (data not shown). In contrast, HEK293 cells showed a weak fluorescence, both in the presence and absence of 10% FCS, but the signal was low compared to the signal for the positive control MessengerMAX (Fig. 8). A quantitative comparison of the average fluorescent intensity (using the nontreated cells as blank) indicated that the fluorescence due to EGFP expression for the cells treated with the VLPs was typically a few times that of the cells treated with bare mRNA, too low to be quantified precisely (Fig. 8d).

Confocal fluorescence microscopy of transfected HEK cells. Cells were incubated for 48 h with 150 ng of mRNA in the presence of 10% FCS.
Cell viability and hemolyticity
Finally, we tested the effect of the VLPs on the viability of HeLa cells and their potential hemolytic activity. At the concentrations used for transfection, the impact on cell viability of HeLa cells, as assessed using a resazurin assay, was negligible. Also, at concentrations that were higher by an order of magnitude, no toxicity was observed for the VLPs (Fig. 9). For the VLPs, we found a dose-dependent increase in the hemolytic activity (Fig. 9). However, the total (extrapolated) hemolysis at an mRNA concentration of 25 ng/μL, a concentration that is two orders of magnitude larger than those used for the transfections in this study, was still less than 25% and is therefore within an acceptable range.
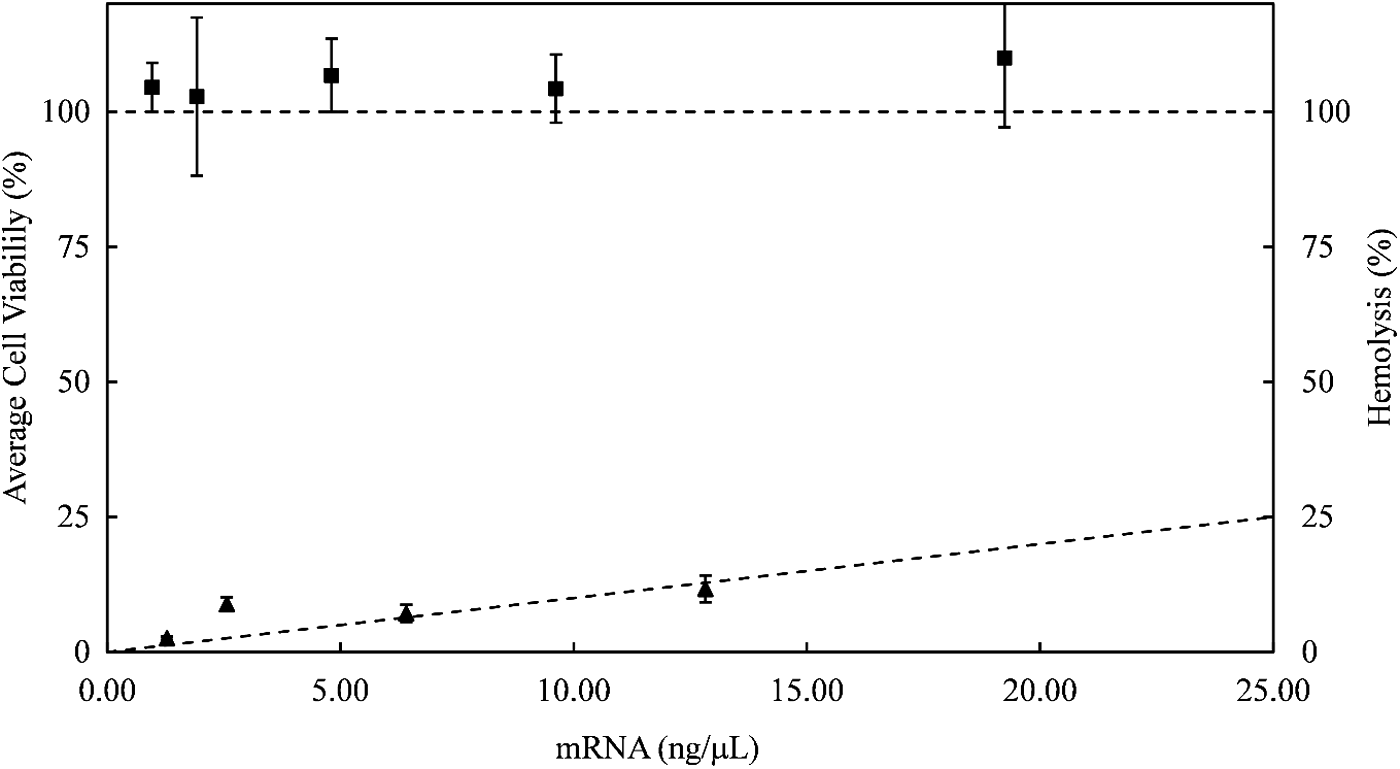
Cytotoxicity to HeLa cells and hemolytic activity of mRNA VLPs at N/P = 3. Square symbols: resazurin assay of average viability of HeLa cells after exposure to mRNA VLPs (filled squares) as a function of mRNA concentration. Filled triangular symbols: percentage of dead red blood cells as a function of mRNA concentration, for mRNA VLPs. Lines through the data points are guides to the eye. Error bars are standard deviations for replicates of the assays.
Discussion
As demonstrated here, the validity of the concept of self-organized VLPs using a simple triblock protein architecture also extends to the packaging and delivery of mRNA. While for linear dsDNA it was shown that each VLP strictly contained one dsDNA molecule [7], for the case of a 996 nt EGFP mRNA, VLPs were rather polydisperse, and presumably contained between one and five mRNA molecules. A possible explanation for why multiple mRNAs are encapsulated in each VLP is mRNA–mRNA interactions that could lead to clusters of mRNA being encapsulated rather than individual molecules. It would be interesting to establish assembly conditions that lead to exactly one mRNA per VLP, as we have done for pDNA, or to assess whether mRNAs that can engage in extensive intramolecular base pairing form more complex structures.
A key difference of the VLPs in comparison to lipoplexes formed by complexation with standard transfection reagents such as Lipofectamine is the very defined molecular architecture of the VLPs. Lipoplexes are molecularly disordered, polydisperse, and most likely a significant fraction of oligonucleotide is exposed at the surface. In contrast, in the VLPs, the mRNA is packaged in the core of the particle. Consistent with the virus-mimicking mode of mRNA packaging, the mRNA was highly resistant against RNases, a major concern in the use of mRNA as a therapeutic. In the same way, this encapsulation should also reduce the immunogenicity of the mRNA.
Moreover, due to the triblock arrangement of functional domains, no positive charges are exposed at the outside of the particle: previously we showed that VLPs encapsulating DNA had a weakly negative zeta-potential, of around −5 mV [7]. The polycationic nature of transfection reagents has been associated with their toxicity. As shown in this study, the VLPs caused no cytotoxicity or hemolysis, also at very high concentrations.
The transfection efficiency of this first-generation mRNA VLPs is still rather low, and sensitive to the presence of serum proteins. This may be attributed to the fact that to this point, the VLPs were designed for encapsulation and no functionalities for cellular targeting and endosomal release have been incorporated, so far. Since the present generation of VLPs is weakly negatively charged and has a very hydrophilic polypeptide brush on its outside, one would not expect these particles to be rapidly taken up by cells, other than by mechanisms that do not involve binding of particles to cells such as macropinocytosis.
Given the observed stability of the VLPs in serum, it does not seem that the serum dependence of transfection is due to either serum-induced disassembly or due to serum-induced aggregation of the VLPs. Possibly, interactions between the serum proteins and the outside of the VLP influence the endocytosis mechanism that is responsible for the uptake of the VLPs.
Therefore, a first modification to be made to the capsid proteins is the addition of a cell-targeting functionality, such that the VLPs bind to cells and possibly activate a more efficient endocytosis pathway. If such targeted uptake can be achieved, the low nonspecific uptake of the mRNA VLPs will be an advantage since unspecific delivery is a shortcoming of all positively charged delivery vehicles. Further possible improvements are to also implement functionalities that mediate endosomal escape [11–13], thus even more closely mimicking the multifunctionality of viruses [14]. The implementation of these functionalities will benefit greatly from the precisely defined architecture of the coat proteins in the VLPs.
Even if cell-targeting functionalities are being used, the interactions of the VLPs with serum proteins will remain a factor contributing to the colloidal stability and uptake of the VLPs. The interactions with serum proteins are determined by the sequence of the outer “C” block, which acts as a shielding domain. Therefore, it will be interesting to explore other shielding polypeptide domains [15] such as elastin-like polypeptides [16,17], PAS-polypeptides [18], and XTEN-polypeptides [19].
A concern for any protein-based nanomedicine is their potential immunogenicity. For the “C”-block used as shielding block of the VLPs in this study, we do not yet have information on its possible immunogenicity. Much data, however, are available for elastin-like polypeptides, dating back to the early article of Urry et al. [20]. Some data on immunogenicity are also available for the PAS and XTEN polypeptide sequences [18,19]. In general, immune responses to these types of disordered peptides are found to be minimal.
In conclusion, the next step will be the implementation of targeting ligands to endow the current particles, tailored for the encapsulation and shielding of mRNA with the capacity for efficient cellular uptake and endosomal release. The modular nature of the protein building blocks provides the basis for rational incorporation and testing of these different functionalities.
Footnotes
Acknowledgments
O.P.S.F. was funded by CAPES Foundation (Science without Borders) grant no. 13173-13-8.
Author Disclosure Statement
No competing financial interests exist.