
Abstract
Antisense oligonucleotides (ASOs) are widely accepted therapeutic agents that suppress RNA transcription. While the majority of ASOs are well tolerated in vivo, few sequences trigger inflammatory responses in absence of conventional CpG motifs. In this study, we identified non-CpG oligodeoxy-nucleotide (ODN) capable of triggering an inflammatory response resulting in B cell and macrophage activation in a MyD88- and TLR9-dependent manner. In addition, we found the receptor for advance glycation end product (RAGE) receptor to be involved in the initiation of inflammatory response to suboptimal concentrations of both CpG- and non-CpG-containing ODNs. In contrast, dosing RAGE KO mice with high doses of CpG or non-CpG ODNs lead to a stronger inflammatory response than observed in wild-type mice. Together, our data provide a previously uncharacterized in vivo mechanism contingent on ODN-administered dose, where TLR9 governs the primary response and RAGE plays a distinct and cooperative function in providing a pivotal role in balancing the immune response.
Introduction
A
As a class, phosphorothioate ASOs have the potential to elicit nonspecific and proinflammatory effects whose severities vary in a sequence-specific manner [2,3]. To circumvent these effects, modification of the DNA using 2′-O-methoxyethyl (MOE) reduced the proinflammatory effects and/or toxicity effect of ASOs [4–9]. As therapeutic candidates, ASOs are carefully designed to have maximal efficacy with minimal to no inflammatory effects in animals. Although most 2′-MOE ASOs fall into this category, a small fraction of sequences examined tend to produce a higher level of proinflammatory effects [3]. While such compounds are identified and removed in preclinical safety studies and will not enter clinical development, a clear understanding of their mechanism of action is warranted.
There is a growing network of receptors and coreceptor involved in the recognition of both DNA and RNA ODNs. Extracellular and cytoplasmic pattern recognition receptors (PRRs) have been described to respond to pathogen-associated molecular patterns and host-derived damage-associated molecular patterns [10,11]. One such family includes the cell surface and endosome-associated toll-like receptors (TLRs) of which TLR3, TLR7, TLR8, and TLR9 recognize nucleic acids [11–14].
The best characterized example of a sequence-specific ODN motif that determines the potency of proinflammatory effects is TLR9 ligands in which ODNs possess optimal unmethylated cytosine-guanosine (CpG) motifs [15–17]. These unmethylated CpG motifs are relatively common in bacteria and DNA viruses, but mainly suppressed and methylated in vertebrate genomes. The localization of TLR9 to the endosomal compartment allows efficient detection of foreign material, while preventing unwanted detection of self-DNA [18,19]. Early structure–activity relationship (SAR) studies demonstrated GACGTT and GTCGTT to be the optimal sequence recognition by mouse and human TLR9, respectively [17,20].
TLR9 ligand receptor-mediated binding engages myeloid differentiation of primary response gene 88 (MyD88) adaptor molecule and activation of mitogen-activated protein kinase family members, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and interferon regulatory factor 7 (IRF7) signaling pathways [14,21,22]. Aside from CpG islands, TLR9-mediated immunostimulatory effects, ODN backbone can also have TLR9 and MyD88-independent inflammatory and/or toxicities [9,23].
More recently, receptor for advance glycation end product (RAGE) was demonstrated to play an important role in promoting lung inflammatory responses to CpG-containing ODN [24]. Using cocrystal structure, Sirois et al. demonstrated that RAGE binds to nucleic acids (both DNA and RNA) by interaction with the charged sugar-phosphate backbones, such as phosphorothioate backbones, of ODN in a sequence-independent manner [24]. This interaction, occurring at the cell surface, promotes endosomal uptake, interaction with TLR9, and subsequent NF-κB activation [24].
In this study, we demonstrate that non-CpG ODN ION-518477 can activate splenic B cells and macrophage populations in a MyD88- and TLR9-dependent manner. Moreover, we show evidence that RAGE receptor is involved in mediating inflammatory response to systemic administration of CpG- and non-CpG-containing sequences. TLR9 governs the primary response observed as an on/off switch and RAGE plays a distinct function by providing a pivotal role in balancing the immune responses to DNA. In addition, we demonstrate that upon ODN challenge, RAGE is differentially regulated in various organs, contributing to organ-specific inflammatory responses to both CpG and non-CpG ODNs.
Materials and Methods
Mice
Animal experiments were approved by the Animal Welfare Committee and conducted according the guidelines of the American Association for the Accreditation of Laboratory Animal Care. All animals were housed in temperature-controlled conditions under a light/dark photocycle with food and water supplied ad libitum. Animal weights were monitored before dosing throughout the live phase of the study. Male Balb/c, C57BL/6 mice were obtained from either Charles River Laboratories or Jackson laboratories.
Breeding pairs for TLR-9- and MyD88-deficient mice in C57BL/6 background were graciously donated by Dr. H. Hemmi (Department of Host Defense, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan) and bred at Charles River Laboratories (San Diego, CA) [25,26]. Male IL1R1- [27] and Male STAT1-deficient mice [28] and their congenic wild-type (WT) control (129SvEv) mice were obtained from Jackson Laboratories and Taconic, respectively. Rage KO mice were obtained from Dr. Esko with permission from DKFZ and were derived as previously described [29], and bred at Charles River Laboratories (San Diego, CA). Scid and TMEM (Sting) were obtained from Jackson laboratories.
ODN design and synthesis
All oligonucleotides were designed and synthesized at IONIA Pharmaceuticals. To identify mouse ASO inhibitors, rapid throughput screens were performed in vitro as previously described [30]. The first five bases and last five bases of chimeric ASO have a 2′-O-(2-methoxy)-ethyl modification, and the ASOs also have a phosphorothioate backbone. ASOs were diluted in phosphate-buffered saline (PBS) for both in vivo and in vitro usage. ODN1826 was purchased from Invivogen and diluted in PBS for both in vivo and in vitro usage.
In vivo treatment with ODN
Compounds were dissolved in PBS at varying concentrations as indicated in the Results section and figure legends (ie, 150 mg/kg), filtered, sterilized, and administered at 10 μL/g animal weight. Immediately before sacrifice, mice were anesthetized with isoflurane and terminal bleed was performed by cardiac puncture. Plasma was isolated from whole blood and analyzed for clinical chemistries and cytokine levels. ALT and AST levels were determined using an Olympus AU400e bioanalyzer. Various organs were collected for flow cytometry, histology, RNA, and protein analysis.
Splenocyte single-cell preparation
Splenocytes were isolated from mice and preparation of single-cell suspension was accomplished by mechanical dissociation using the gentleMACS Octo Dissociator (Miltenyi Biotech, Inc.). Briefly, individual spleens were transferred to gentleMACS C tube containing 5 mL of PBS. Spleens were dissociated using program m_spleen_01. Cell suspension was passed through a cell strainer and spun for 5 min at 1,250 rpm. Red blood cells were lysed as per manufacturer's instructions (eBioscience). Cells were passed through a cell strainer and spun for 5 min at 1,250 rpm. Cells were counted and either used for flow cytometry or placed in culture for ex vivo treatment; in some instances, splenocytes were resuspended in the MACS buffer for magnetic cell purification as per manufacturer's instruction (Myltenyi Biotec, Inc.).
Isolation of peripheral blood mononuclear cells using Ficoll-Paque
Briefly, a buffy coat was obtained and diluted into PBS containing 2 mM EDTA and layered over 15 mL of Ficoll-Paque in a 50 mL conical tube. Tubes were centrifuged at 400g for 30 min at 20°C (no brakes). Upper layer was aspirated without disrupting the interphase containing mononuclear cell layer. The interphase was carefully transferred to a new 50 mL conical tube and diluted in the wash buffer followed by centrifugation at 300g for 10 min at 20°C (no brakes). Supernatant was carefully removed and cells were resuspended in the MACS buffer for magnetic cell separation as per manufacturer's directions (Miltenyi).
Whole blood assay
Whole blood was obtained from naive animals by cardiac puncture into EDTA collection tubes. Whole blood collected by cardiac puncture was diluted 1:1 in RPMI media containing 10% fetal bovine serum (FBS), stimulated with either PBS (saline), ION-518477 or ODN1826 were indicated for 24 h. Supernatant was collected and frozen for further analysis.
Flow cytometry
Cell suspensions were as described above and prepared by sieving and gentle pipetting. After washing with ice-cold buffer (0.5% BSA in PBS), the cells were incubated with antibodies for a minimum of 20 min and washed thrice with FACS buffer. Cells were fixed using BD fixing solution (BD Biosciences) and kept in the dark at 4°C for next day analysis.
RNA isolation and qPCR analysis
RNA was extracted from liver samples using RNeasy columns (Invitrogen) according to manufacturer's protocol. RNA samples were analyzed by fluorescence-based quantitative real-time polymerase chain reaction using an Applied Biosystems 7700 sequence detector. Target RNA levels were normalized to the total RNA concentration determined using RiboGreen or to housekeeping gene Gapdh. Primers and probes for analysis of the expression of different genes were either designed using Primer Express Software (Applied Biosciences) or purchased from Invitrogen. Quantitative polymerase chain reaction (qPCR) was performed using 50 ng of total RNA.
Meso Scale Discovery platform
Mouse blood was obtained by cardiac puncture and plasma was collected by centrifugation for 10 min at 4°C at 10,000 rpm. Plasma and supernatant collected from cultured cells were frozen at −20°C for further use; 25 μL was used for Meso Scale Discovery as per manufacturer's recommendation (MSD).
Statistical analysis
The data of in vivo studies (at least four animals per group) are expressed as the mean ± standard deviation and were analyzed by ANOVA. When significance was obtained, multiple comparison analysis was conducted using Bonferroni as a post hoc test. In some instances, unpaired Student's t-test was utilized. All graphs and statistical analysis were obtained using Graph Prism (Graphpad Software). Flow cytometry analysis was obtained using FACSDiva v7 (BD Biosciences).
Results
Non-CpG ODN induces an MyD88- and TLR9-dependent immune response in vivo
ION-421856 was identified as inflammatory during a routine screen to identify potent 2′-MOE ASO ODNs capable of silencing the mouse B4galt6 transcript. We ruled out the possibility that ION-421856-mediated inflammatory response was due to B4galt6 knockdown (data not shown). In vivo, ION-421856 was readily detectable as proinflammatory for its ability to induce 3–5-fold spleen weight increase in either Balb/c or C57BL/6 mice treated subcutaneously (s.c.) with 100 mg/kg/week for 6 weeks (Fig. 1a, left panel). ASOs have been shown to accumulate in the liver and for this reason, we monitor liver weight and serum chemistries for signs of hepatotoxicities. Liver weight increase was more significant in Balb/c strain than C57BL/6-treated mice (Fig. 1a, right panel) with no significant elevation in blood alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (data not shown), indicating that ION-421856 possesses proinflammatory properties, but no significant hepatotoxicity.
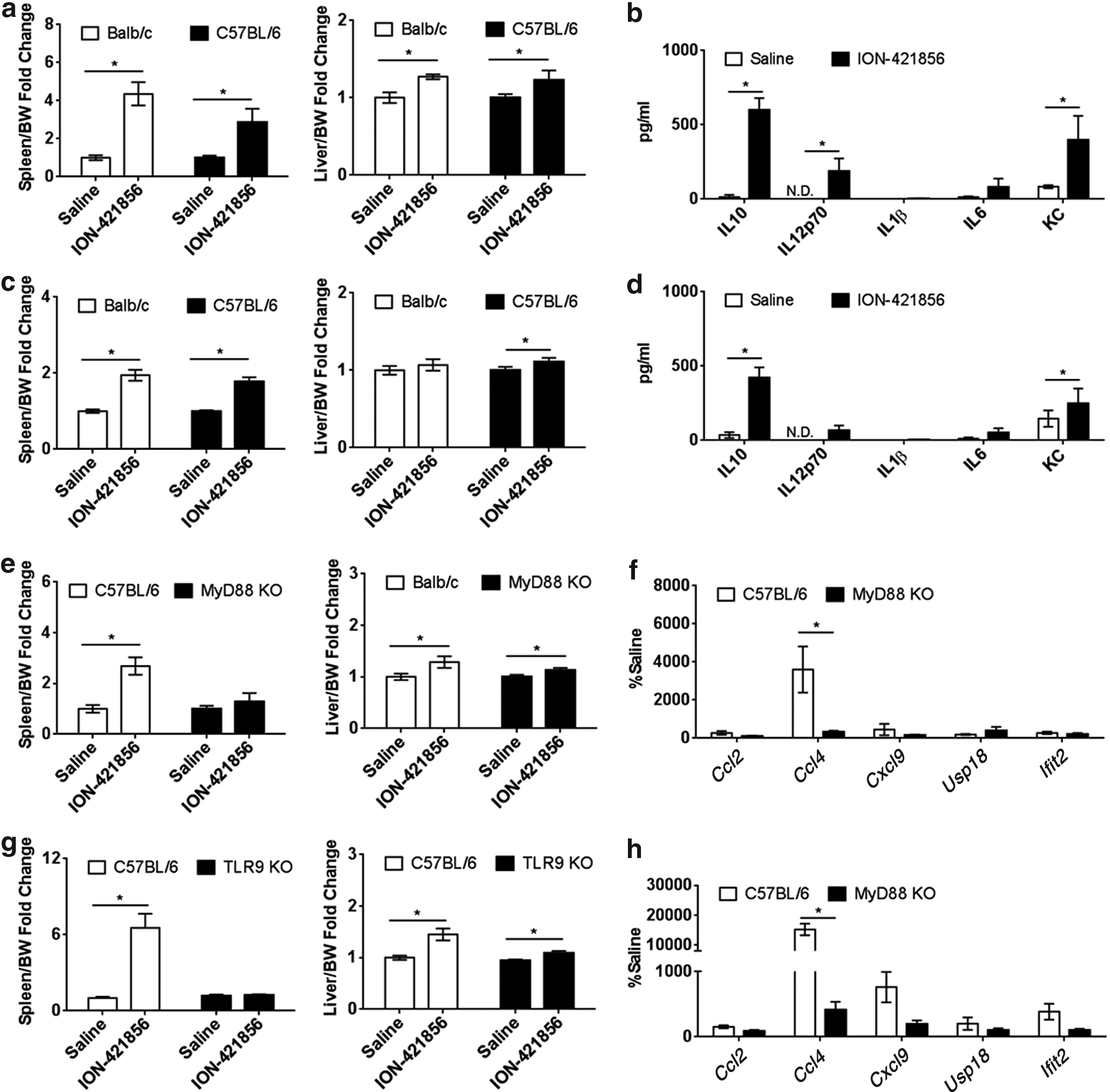
ION-421856 exerts its proinflammatory properties in a MyD88-TLR9-dependent manner.
Substantial increase in circulating levels of inflammatory markers such as interleukin (IL)-16, IL10, IL12p70, and keratinocyte chemoattractant (KC; also known as CXCL1) was detected in the plasma of treated C57BL/6 animals (Fig. 1b), but no detectable IL1β production was observed. This was not a chronic phenomenon since the same proinflammatory effects were observed at 72 h following a single dose (300 mg/kg) (Fig. 1c, d). Spleen weight (twofold) was significantly increased in both animal strains (Fig. 1c, left panel) with no obvious signs of hepatotoxicity (Fig. 1c, right panel). Increased circulating levels of IL10, IL12p70, IL6, and KC were also detectable following acute dosing (Fig. 1d).
The induction of inflammatory cytokine was an unexpected finding since non-CpG 2′-MOE ASOs generally do not elicit a strong inflammatory response due to their lack of immunostimulatory motifs. We reasoned that dissecting the molecular mechanism involved in these unusual immunostimulatory responses would provide insight allowing us to avoid this effect in future drug candidates.
To dissect factors involved in the mechanism of action, the following mouse knockout (KO) models were utilized: IL1R, Caspase 1, cluster of differentiation (CD14), stimulator of interferon genes (Sting), signal transducers and activators of transcription STAT1, MyD88, and TLR9. Lack of proinflammatory effects in the KO models would indicate an important component of ION-421856-mediated inflammatory responses. Spleen weights were recorded as an indication of inflammation. All corresponding WT animals demonstrated a spleen weight increase following treatment with ION-421856 in comparison to saline-treated groups (Fig. 1e, g and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat). Splenomegaly was also observed in IL1R, Casp1, CD14, and Sting KO, with no significant difference from the WT counterpart, indicating that these signaling pathways are not, or minimally involved in ION-421856-mediated inflammatory responses (Supplementary Fig. S1a–d).
In contrast, splenomegaly was significantly abrogated in STAT1 (Supplementary Fig. S2a), MyD88 (Fig. 1e), and TLR9 KO (Fig. 1g) mice when compared to WT-treated mice; with minimal changes in liver weights (Fig. 1e, g, right panel; Supplementary Fig. S2b). In addition, liver mRNA induction of Ccl2, Ccl4, Cxcl9, Usp18, and Ifit2 were all abrogated in STAT1 (Supplementary Fig. S2c), MyD88, and TLR9 KO models (Fig. 1f, h), further supporting a diminished proinflammatory response. Also, we show that ION-421856 causes an increase in inflammatory infiltrates as demonstrated by histological examination using anti-CD68 and anti-F4/80, and anti-Ly6C antibodies of stained livers from mice (Supplementary Fig. S2d, e) with fewer cell infiltrates in liver tissue from both STAT1 and MyD88 KO mice. Together, these data demonstrate that non-CpG ASO ION-421856 can effectively trigger an immune response in a TLR9/MyD88, STAT1-dependent manner in vivo.
Influence of length and sequence on inflammatory response
We performed a SAR analysis to better understand the proinflammatory properties of ION-421856 (Fig. 2). ION-421856 is a 20 nucleotide oligo with a phosphorothioate backbone where the first and last five bases have a 2′-MOE modification (chemistry referred in Fig. 2a table as 5-10-5 MOE). Increasing the length from 5-10-5 MOE to 7-10-7 MOE (ION-518476) completely abrogated the proinflammatory properties of ION-421856 (Fig. 2a). On the other hand, reducing the length of the 5′-end from 5-10-5 MOE to 4-10-X MOE (where X = 3, 4, or 5) led to a significant increase in spleen weight fold change [ION-549905 (5-fold), ION-518477 (10-fold), and ION-549902 (7-fold)] (Fig. 2a). ION-518477 was the most potent oligo to induce splenomegaly in Balb/c (6-week study) (Fig. 2a). At 96 h, following a single-dose administration of ION-518477, we observed a threefold increase in spleen weight compared to 1.5-fold increase with ION-421856 (Fig. 2b).
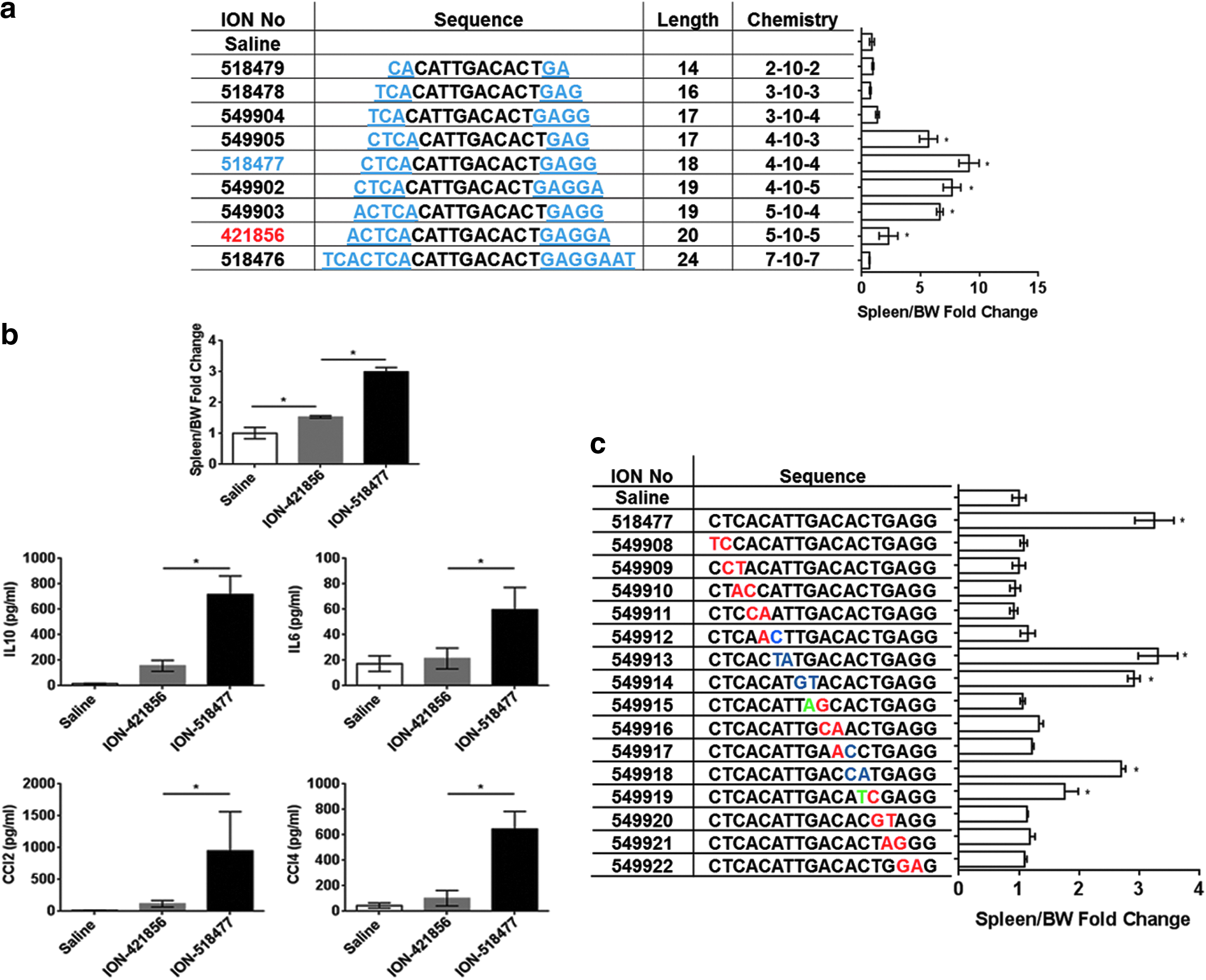
Identification of ION-518477
In addition to spleen weight increase, we monitored various cytokines and chemokines circulating in the plasma of animals. Consistently, ION-518477 led to the production of IL6, IL10, CCL2, and CCL4, demonstrating a strong systemic response following a single-dose treatment with ION-518477, which was significantly higher than ION-421856 (Fig. 2b).
Furthermore, spleen weight increase was also observed when shortening the 3′-end from 5-10-5 MOE to 5-10-4 MOE (ION-549903) (Fig. 2a). However, splenomegaly was completely abrogated when the 5′-end was further truncated to a 3-10-X (where X = 3 or 4) MOE (ION-518478 and ION-549904; Fig. 2a). Reducing the length of both ends to 2-10-2 MOE also resulted in no spleen weight increase (ION-518479; Fig. 2a). Removal of the terminal 5′end cytosine appeared to be important for eliciting a stronger immune response (compare ION-549905 and ION-549904; Fig. 2a).
The influence of sequence on the potency of inflammatory effect for ION-518477 was investigated by systemically inverting dinucleotides throughout the ASO or making selected base substitutions. Interestingly, while the terminal 5′ cytosine base appeared to be important for immune modulation, nucleotide permutations in the gap sequence could also influence the inflammatory responses, exemplified by modifications of ION-518477 (Fig. 2c), suggesting both the sequence and likely secondary structure, in addition to the length of the MOE modified sequence are important parameters influencing immune response by non-CpG ODNs. Whether this would classify non-CpG ODNs into various subclasses as observed with CpG-containing ODNs remains to be elucidated. Because of its strong proinflammatory properties, ION-518477 was utilized to further dissect the molecular signaling involved in non-CpG-mediated inflammatory responses.
ION-518477 elicits a strong immune response in vivo
Next, we sought to investigate the in vivo and in vitro proinflammatory properties of ION-518477. First, we tested various routes of administration in C57BL/6 mice and compared spleen weight and circulating cytokines and chemokines in the plasma of treated animals. The same dose (150 mg/kg) was administered either s.c., i.p. (intraperitoneal), or i.v. (intravenous) for 24 h. All endpoints tested clearly demonstrated that the route of administration results in a similar spleen weight increase (Fig. 3a) and cytokine/chemokine profile (Fig. 3b) with the exception of CCL4, which was significantly increased in i.p. administration and further augmented through the i.v. route of administration (Fig. 3b). We concluded that the route of administration does not significantly alter the inflammatory profile of ION-518477 in vivo.
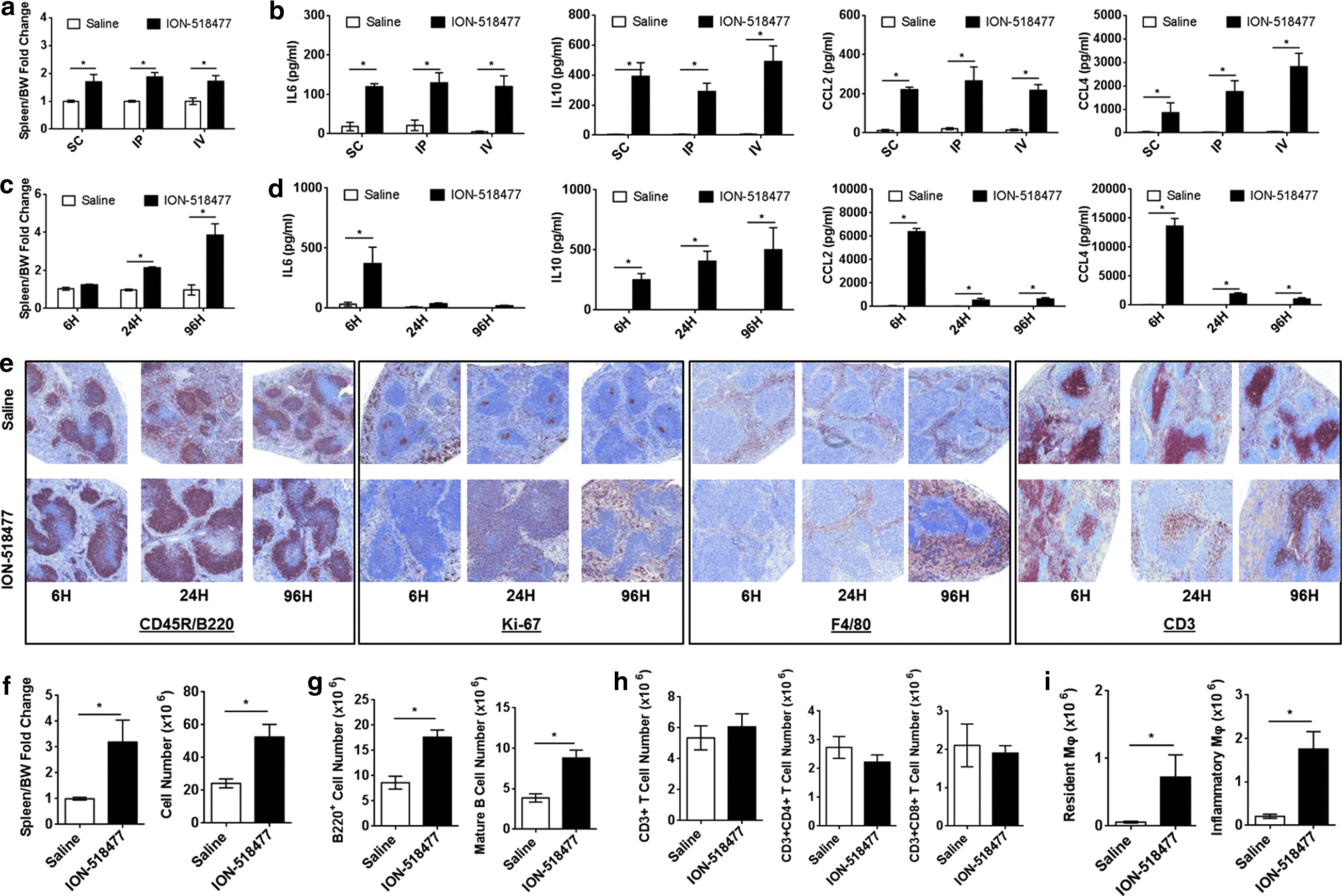
ION-518477 elicits a strong immune response in vivo.
Subsequently, we carried out a time-course study where animals were dosed subcutaneously and sacrificed at 6, 24, and 96 h postchallenge at 150 mg/kg. Splenomegaly was recorded as early as 6 h and steadily increased from 24 to 96 h (Fig. 3c). Interestingly, IL6, CCL2, and CCL4 production peaked at 6 h and gradually decreased over time, whereas IL10 steadily increased over time and correlated with spleen weight increase (Fig. 3d). Immunostaining for CD45R/B220 revealed splenic B cell hyperplasia peaking at 24 h in animals treated with ION-518477 (Fig. 3e) with Ki-67-positive cells observed in the germinal center and red pulp at 24 h, and increasing by 96 h (Fig. 3e). We also show that F4/80+ macrophages are significantly augmented in the red pulp at 96 h (Fig. 3e). We also monitored CD3+ cells and revealed that T cell staining pattern decreased by 24 h, but was restored by 96 h, a phenomenon that we did not further investigate (Fig. 3e).
These findings clearly indicate that ION-518477 causes a shift in cellular composition of the spleen that appears to be largely governed by changes in B cells and macrophages. These results prompted us to perform multiparametric analysis of spleens by utilizing flow cytometry. An increase in spleen cell count (Fig. 3f, right panel) occurred concomitant with a fourfold spleen weight increase (Fig. 3f, left panel) at 96 h. B220+ B cell population was significantly increased in comparison to saline-treated group (Fig. 3g, left panel), confirming our immunohistochemistry data. The increase in B cell population was driven by a change in the mature B cell population (Fig. 3g, right panel), and not marginal, transitional (T1 and T2), or immature B cells (Supplementary Fig. S3a–d), which also corroborated our histological finding of increased B220+ cells in germinal centers where proliferation and differentiation occur. T cell subsets were separated by CD4 and CD8 surface expression, demonstrating no changes in T cell populations at 96 h (Fig. 3h) [31]. In addition, there was a significant increase in resident and inflammatory macrophages (Fig. 3i) [32], thus confirming that B cells and macrophages play a role in ION-518477-mediated inflammatory responses in vivo.
ION-518477 elicits a strong immune response in vitro
To demonstrate direct B cell and macrophage activation, we utilized freshly isolated splenocytes and treated them ex vivo with ION-518477. The ex vivo kinetics of cytokine response was different than that observed in our in vivo studies. IL6, IL10, and CCL4 were all steadily increased over time, with a slight increase in CCL2 (Fig. 4a). The production of cytokines and chemokines was shown to be dose dependent at 24 h (Fig. 4b) with a biphasic response in cytokine production at a very high dose due to an increase in cellular stress (data not shown).
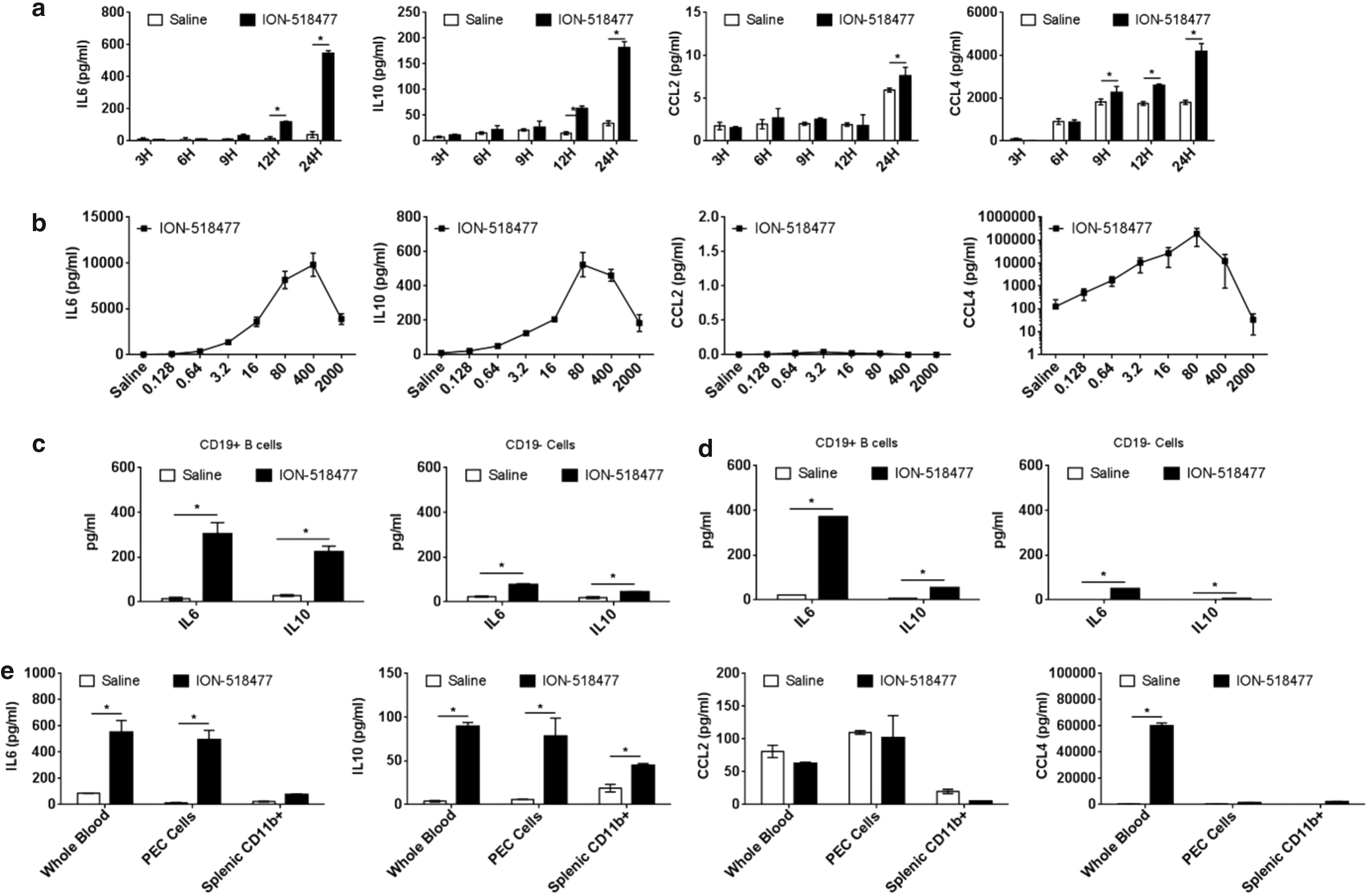
ION-518477 elicits a strong immune response ex vivo.
Stimulation of magnetically purified CD19+ splenic B cells resulted in a larger production of cytokine release (IL6 and IL10) than the non-B cell (CD19−) population (Fig. 4c). Interestingly, this observation was also observed with fractionated human PBMCs (Fig. 4d), indicating that B cells must play a very important role in the production of IL6 and IL10 in response to ION-518477. These results also indicated that ION-518477 is a cross-species immunostimulatory compound. Using flow cytometry analysis of splenocytes, we demonstrate an increase in B220+ cell population and activation as monitored by the upregulation of CD69 (Supplementary Fig. S4a), with no clear changes in macrophage populations (Supplementary Fig. S4b), which did not correlate with our in vivo findings demonstrating a clear increase in both resident and inflammatory macrophages in the spleen of treated mice (Fig. 3i).
This discrepancy led us to believe that cellular activation could possibly occur outside the spleen. To test this hypothesis, we compared the ability of whole blood, thioglycollate-induced peritoneal exudate cells (PEC), and splenic CD11b+ cells to respond to stimulation ex vivo (Fig. 4e). Interestingly, IL6 and IL10 levels were similarly produced in either whole blood or PEC cells, which are rich in monocytes/macrophages, whereas splenic CD11b+ cells did not respond as well (Fig. 4e). Thus, circulating cells in whole blood produced much higher levels of CCL4 than PECs or splenic CD11b+, indicating that circulating cells, including monocytes/macrophages, can directly respond to ION-518477 by secreting cytokines and chemokines, possibly resulting in cells homing to secondary lymphoid organs such as the spleen where they differentiate and become activated.
ION-518477-mediated inflammatory response can occur in the absence of B cells
To obtain a better understanding of the in vivo relationship between macrophages and B cells in the initiation and propagation of inflammatory responses, we dosed severe combined immune deficiency mutation (Scid) mice with ION-518477 at 150 mg/kg for 96 h. Body weights were similar across all groups tested (Fig. 5a). Basal, untreated spleen weights were initially much smaller in Scid mice compared to WT (Fig. 5b), characterized by a lack of functional B and T cells in this model. Following treatment with ION-518477, spleen weight fold change observed in Scid mice was exacerbated in comparison to WT-treated mice (Fig. 5c, left panel) and this exacerbation was greater when comparing splenocyte cell count (Fig. 5c, right panel). This exaggerated response correlated with the increase in resident and inflammatory macrophage populations (Fig. 5d).
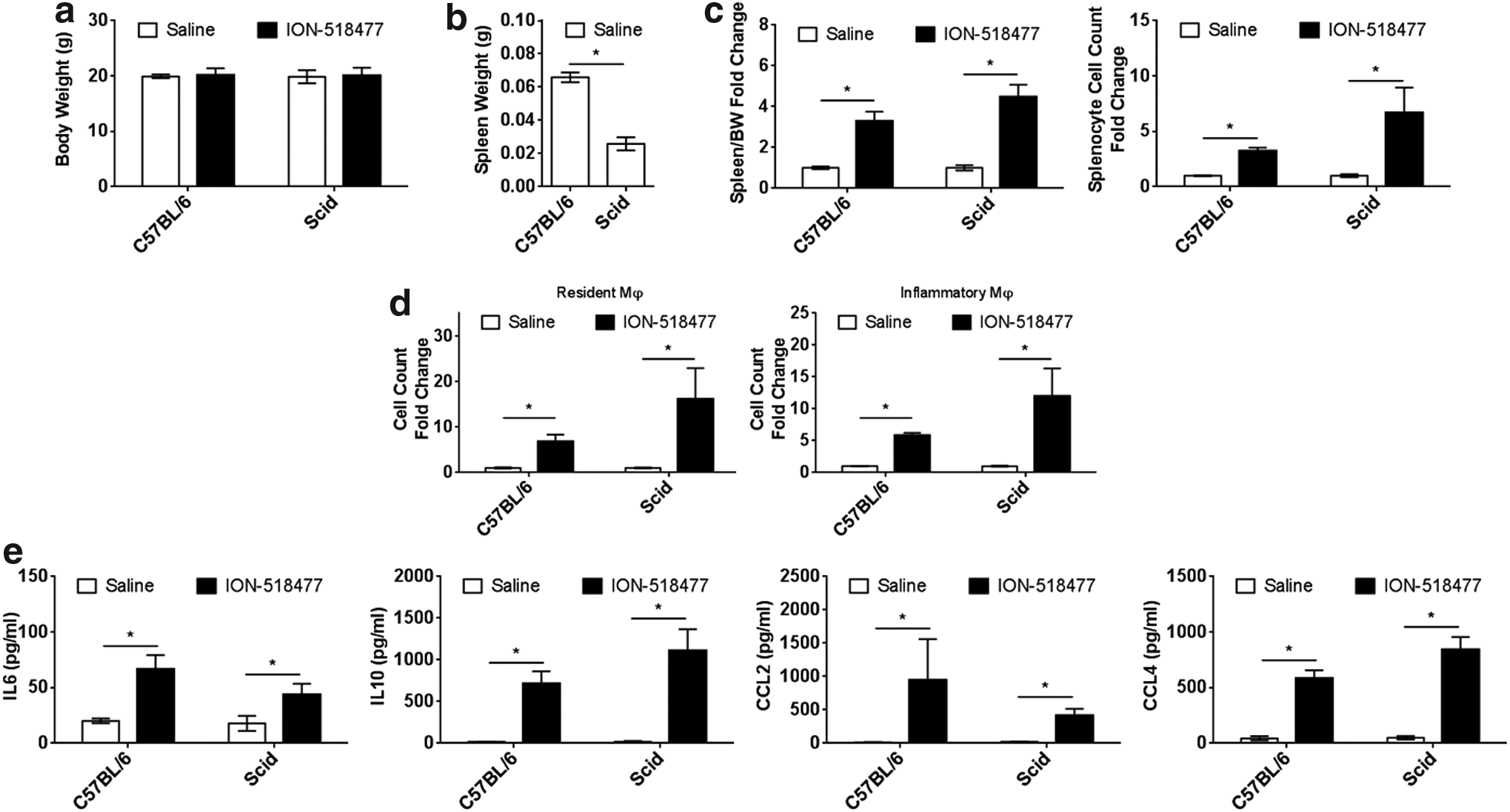
ION-518477-mediated inflammatory response can occur in the absence of B cells.
Levels of circulating IL6, IL10, CCL2, and CCL4 were all readily detectable in WT and Scid mice, indicating that under these conditions, B cells and T cells do not play a contributing role in their production (Fig. 5d). The Scid model highlights that non-CpG ASO can mediate an inflammatory response independent of mature B cells.
ION-518477 signals through TLR9 receptor and MyD88 adaptor molecule
MyD88 is an important adaptor molecule found downstream of various PRRs, including TLR9 [12,25]. We showed that parent compound ION-421856 exerts its proinflammatory properties in a MyD88-/TLR9-dependent manner (Fig. 1). Thus, we confirmed the role of MyD88/TLR9 in ION-518477-mediated inflammation. As expected, treatment with ION-518477 induced changes in spleen weight, splenocyte cell count, and chemokine and cytokine production, which were all abrogated in both TLR9 and MyD88 KO models (Fig. 6a, b). At the cellular level, we confirmed that B cell proliferation, as well as increase in resident and inflammatory macrophage cells were also MyD88- and TLR9-dependent events (Fig. 6c). We also confirmed that ex vivo splenocytes treated with ION-518477-mediated inflammatory responses in a MyD88- and TLR9-dependent manner (Fig. 6d). Altogether, these results clearly demonstrate the importance of both MyD88 and TLR9 in mediating inflammatory responses to non-CpG oligonucleotides both in vivo and in vitro.
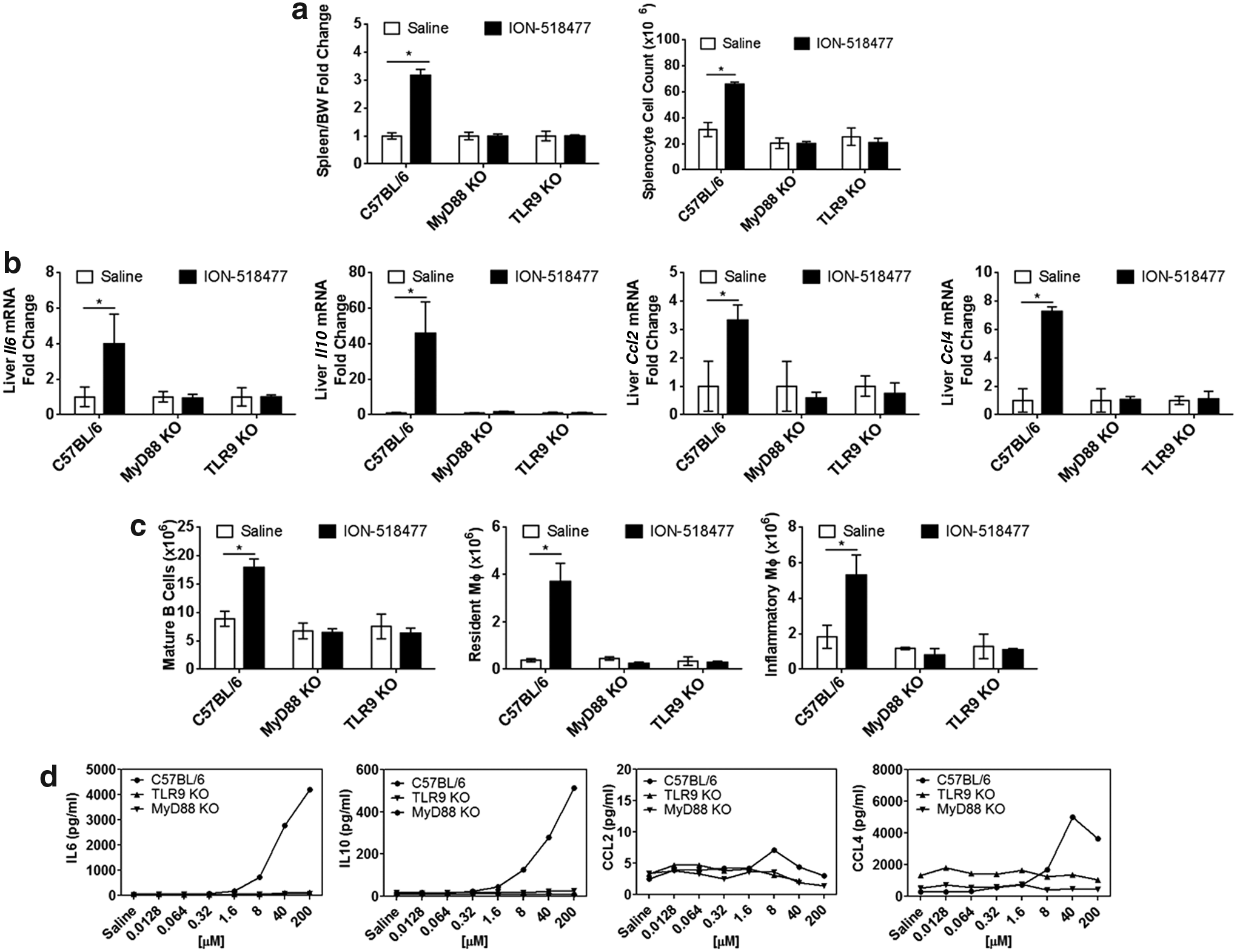
ION-518477 signals through TLR9 receptor and MyD88 adaptor molecule.
RAGE mediates inflammatory responses to low doses of CpG and non-CpG ODNs in vivo
One important feature of ODN recognition by PRRs is the internalization of the nucleic acids within the endosomes to make contact with TLRs. One such molecule recently shown to facilitate CpG-ODN recognition by TLR9 is RAGE [24,33]. Thus, we hypothesized that non-CpG ASOs and RAGE engage in TLR9-mediated inflammatory response in vivo.
The role of RAGE in directly controlling systemic inflammatory responses remains elusive. Sirois et al. administered a low dose of ODN1826 by intranasal administration, demonstrating that RAGE plays an important role in pulmonary inflammation to CpG ODN [24] by showing a reduction in cytokine production in RAGE KO mice [24]. With this in mind, we administered a low dose of ION-518477 or ODN1826 (Fig. 7a, b) subcutaneously to C57BL/6 mice and RAGE KO mice and monitored spleen weight as well as plasma cytokines (IL6 and IL10) and chemokines (CCL2 and CCL4) at 4 h postadministration. We collected plasma at 4 h based on our kinetic study (Fig. 3c), demonstrating peak of cytokine and chemokine production at an early time point following single dosing. Spleen weight increase was not significantly affected at this time point (Fig. 7a). However, circulating levels of IL6, IL10, CCL2, and CCL4 were clearly reduced in the RAGE KO mice in comparison to WT (Fig. 7b). Interestingly, this difference was more significant in animals treated with non-CpG ODN ION-518477 than CpG-containing sequence ODN1826, indicating that, at low doses of administered ODN, RAGE plays a role in mediating immune response to both CpG- and non-CpG-containing ODNs. It is worth noting that in the RAGE, KO production of cytokines and chemokines remained higher than the saline-treated group, indicating that another receptor such as TLR9 is involved in this response.
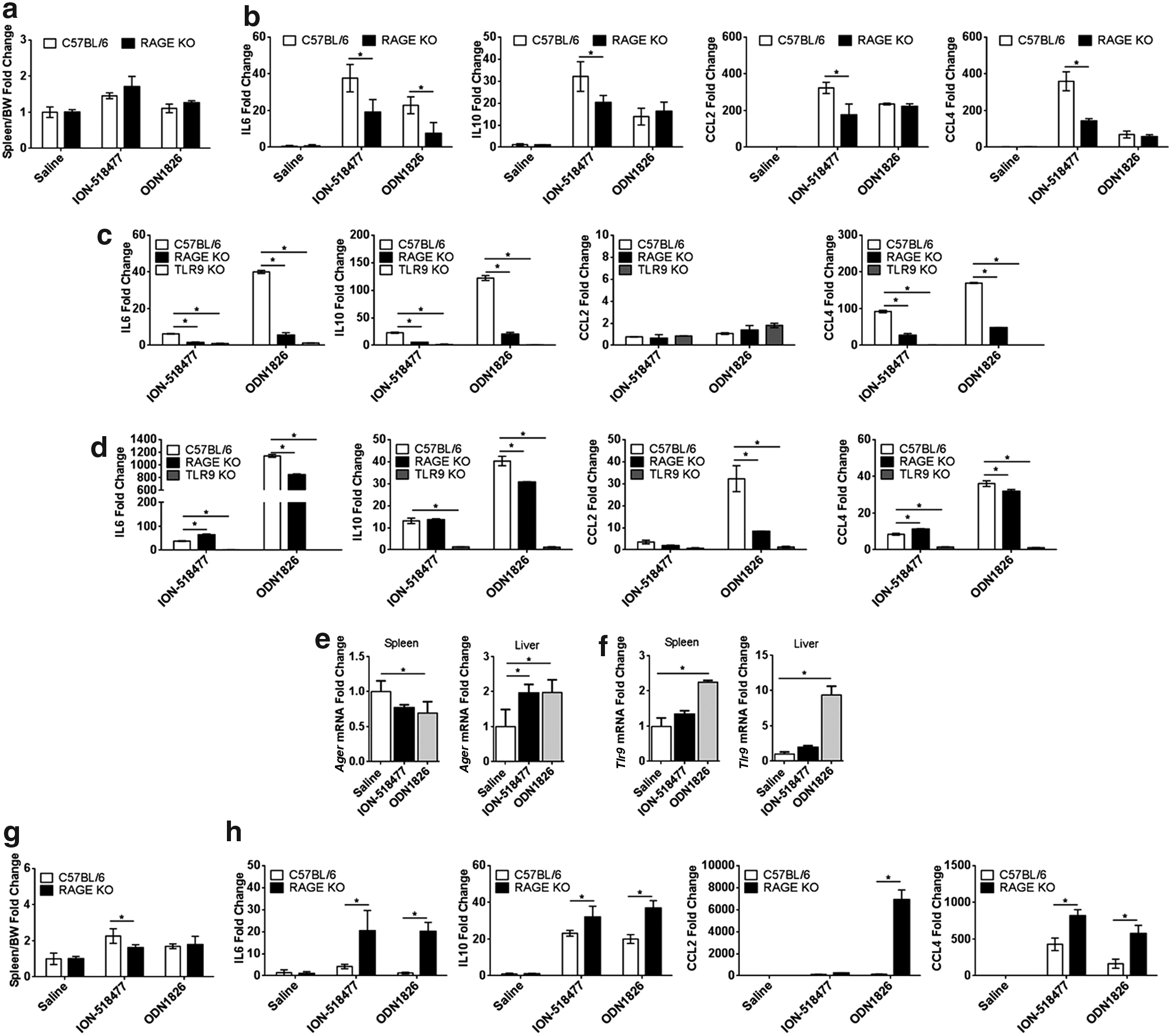
RAGE mediates inflammatory responses to suboptimal CpG and non-CpG ODN concentrations in vivo and in vitro.
To confirm our in vivo results, whole blood from WT, RAGE, and TLR9 KO mice was treated ex vivo with ION-518477 or ODN1826 (Fig. 7c). We observed a significant decrease in IL6, IL10, and CCL4 production in both RAGE and TLR9 KO whole blood in comparison to WT mice for both CpG and non-CpG sequence (Fig. 7c). Altogether, these results indicate that plasma circulating cells employ both RAGE and TLR9 to produce inflammatory cytokines and chemokines. Interestingly, freshly isolated splenocytes from RAGE KO mice, but not TLR9 mice, produced similar or higher levels of IL6 and CCL4 to both CpG and non-CpG ODNs than WT cells, raising the possibility that splenocytes, mainly composed of B cells, signal strictly in a TLR9-dependent manner (Fig. 7d). This prompted us to examine the levels of mRNA for Ager (RAGE) and Tlr9 in the spleen as well as the liver.
We demonstrate that following challenge with ION-518477 and ODN1826, RAGE and TLR9 mRNA levels are differentially regulated in an organ-specific manner (Fig. 7e, left panel). Ager (RAGE) mRNA was decreased in the spleens of treated animals for both ION-518477 and ODN1826 and increased in the liver of treated animals (Fig. 7e, right panel). On the other hand, Tlr9 mRNA expression was consistently increased in both organs (Fig. 7f). These results demonstrate for the first time that upon challenge with either non-CpG or CpG ODN, RAGE and TLR9 are differentially regulated in the spleen and liver of treated animals.
RAGE negatively regulates inflammatory responses to high doses of administered ODNs in vivo
We demonstrate in this study that, RAGE and TLR9 are inducible following CpG and non-CpG ODN in vivo challenge (Fig. 7e, f). Inducibility of RAGE and subsequent signaling events was reported to correlate with ligand accumulation leading to inflammatory diseases and cancers [34]. Thus, we decided to challenge the RAGE KO mice with a higher dose of both ION-518477 and ODN1826. Unexpectedly, at these doses, spleen weight remained largely unchanged by administration with either ION-518477 or ODN1826 in the RAGE KO compared to WT mice (Fig. 7g).
Interestingly, levels of circulating cytokines (IL6 and IL10) and chemokines (CCL2 and CCL4) in the plasma of treated animals at 4 h posttreatment were all significantly higher in the RAGE KO compared to WT-treated mice, indicating that at a higher dose, RAGE plays a distinct role compared with our initial hypothesis. In fact, the increased response points to the possibility that in WT animals, RAGE negatively controls systemic inflammation. Taken together, our results demonstrate for the first time that RAGE plays a role in systemic inflammation and confers a response that is both dependent on cellular composition of organs and ODN dose dependent.
Discussion
CpG- and non-CpG-containing ODN molecules are known to be potent activators of innate immune responses and yet the exact mechanism underlying sensing, internalization, receptor activation, and downstream signaling cascades is not fully understood in vivo. It is well accepted that TLR9 plays an important role in DNA recognition and signaling, particularly in distinguishing DNA molecules harboring CpG motifs [15,35]. However, TLR9 KO mice challenged with certain microbial DNA suggest that other receptors and/or coreceptors are involved in this process [36]. Of interest to our research is the RAGE receptor that can act as a PRR for nucleic acid and aid TLR9 in initiating an immune response [24,33]. Several lines of evidence, including our work, support the importance of RAGE and TLR9 in the acute response to both CpG- and non-CpG-containing ODNs.
Intriguingly, immune activation by low dose of ODN was dependent on the RAGE receptor, whereas high concentrations appeared to bypass the need for RAGE to signal through TLR9. Although not demonstrated in this study, this multivariate mechanism of action could prove to be critical in balancing the presence of self-nucleic acids and plays a determining role in tipping the scale toward an exaggerated response when DNA concentrations are too elevated, leading to chronic inflammation. It will be interesting to study the role of RAGE and TLR9 in chronic challenge by ODN as well as in disease settings such as autoimmunity and cancer, which have implicated both TLR9 and RAGE receptors [34,37].
Our work brings valuable information about the in vivo biology of RAGE in controlling inflammatory responses to non-CpG ODNs and highlights the complexity of this receptor. An interesting aspect of RAGE biology that was not covered in this study and is beyond the scope of this work is the soluble forms that can accumulate through two distinct mechanisms: proteolytic cleavage (sRAGE) by MMP-9 or ADAM10, or alternative mRNA splicing (esRAGE) [38–41]. In either case, the soluble forms are considered to be inhibitory by directly binding and preventing downstream signaling of cell surface RAGE. Its existence points the possible role of RAGE as a negative regulator of inflammation in which binding of circulating RAGE to membrane-bound RAGE would prevent downstream activation, acting as an endogenous protective factor or negative feedback loop mechanism, which raises the possibility that lack of soluble RAGE could have contributed to the KO phenotype we observed when treating mice with higher doses of both CpG and non-CpG ODNs.
Moving forward, there are plenty of questions that remain to be addressed in regard to RAGE, TLR9, and their collaboration and distinct functional roles in controlling immune responses to DNA. Both receptors clearly participate in maintaining a delicate balance between clearance and induction of exaggerated responses. Adding to this multifaceted relationship is their organ-specific involvement and contribution to the onset and progression of inflammatory responses. As we observed in our study, regulation of RAGE appears to be more tissue specific than TLR9 after ODN exposure and points to the possibility that B cells recognize and internalize ODN to assist TLR9 through a different coreceptor than RAGE. Taken together, understanding RAGE and TLR9 biology will be helpful to develop novel ways to manipulate and restore inflammatory diseases, overcome uncontrollable inflammation, and/or stimulate a specific response. Both RAGE and TLR9 are attractive therapeutic molecules for which both agonist and antagonist would prove to be beneficial depending on clinical settings.
Footnotes
Acknowledgments
Breeding pairs for MYD88 KO mice were graciously donated by Dr. H. Hemmi (Department of Host Defense, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan). Authorization to use the RAGE KO mice came from DFKZ (Germany). The authors would also like to thank Dr. Hiroki Kato, Dr. Jordan Pober, Dr. Irina Caminshi, and Dr. Patrik Andersson for helpful discussions. The authors would also like to acknowledge the oligonucleotide synthesis and screening groups at IONIS Pharmaceuticals.
Author Disclosure Statement
All authors, except L.B., are/were employees of IONIS Pharmaceuticals and have a vested financial interest. L.B. was an intern at IONIS and has no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.