
Abstract
This white paper, which is the 10th in a series intended to address issues associated with the development of therapeutic oligonucleotides, examines the subject of product-related impurities. The authors consider chemistry and safety aspects and advance arguments in favor of platform approaches to impurity identification and qualification. Reporting, identification, and qualification thresholds suitable for product-related impurities of therapeutic oligonucleotides are proposed.
Background
S
The encouraging clinical progress and continued high level of interest in oligonucleotide therapeutics have spurred efforts by the oligonucleotide safety working group and others to develop and publish position papers that seek to provide sound scientific advice in a number of areas. The articles aim, at least in part, to fill gaps in scientific advice that arise because oligonucleotide therapeutics occupy a space somewhere between small-molecule drugs and biologics and that, consequently, it is not always possible or appropriate to apply guidelines established for small molecules or biologics. To date, the results of the effort comprise eight position papers that focus on nonclinical issues [1–8] and a ninth, chemistry-related article on the topic of drug substance specifications [9]. In this, the 10th article in the series, the authors attempt to provide scientific advice on the topic of impurities in oligonucleotide drug substances and drug products. The article considers chemistry and safety aspects and seeks to find an effective balance of being helpful for the majority of oligonucleotides without appearing prescriptive. The reader is cautioned that adherence to the advice given in the following sections does not guarantee regulatory agency endorsement of the approaches described. For this reason, sponsors of oligonucleotide therapeutics are strongly encouraged to discuss all questions pertaining to impurity identification, reporting, and qualification with regulatory agencies during the drug development process.
Introduction
The International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidelines Q3A(R2) and Q3B(R2) classify nongenotoxic impurities as organic, inorganic, or residual solvents. Organic impurities are further divided into process- and product-related types. Although precise definitions are not provided, it is generally accepted that process-related organic impurities comprise molecules such as starting materials, ligands, reagents, and protecting groups, whereas product-related organic impurities are molecules more closely structurally related to the active ingredient and formed due to side reactions or on storage (this is consistent with the use of the same terms in ICH Q6B). The current classification system is also appropriate for impurities of oligonucleotide therapeutics. Furthermore, process-related organic impurities (as defined above), inorganic impurities, and residual solvents of oligonucleotide therapeutics are covered adequately by the current ICH small-molecule guidelines. For example, it is clear that ICH Q3C(R6), the guideline that covers residual solvents in small-molecule drugs, can be applied to oligonucleotide therapeutics. Likewise, current guidance on elemental impurities (ICH Q3D) also is directly applicable as written. Because they are addressed adequately by current guidance, process-related organic impurities, inorganic impurities, and residual solvents are not discussed herein; rather the focus of this article is product-related organic impurities. 2
For small-molecule drugs, it is important to address both chemistry and safety aspects of impurities. Chemistry topics include impurity characterization, the reporting of impurities in drug substance and drug product specifications, and the development of appropriate specification limits. From the safety perspective, it is important to consider impurity qualification, that is, the process by which the safety of an impurity is established. Although certain technical challenges exist, the general chemistry and safety principles that apply to impurities of small-molecule drugs are also applicable to product-related impurities of oligonucleotide therapeutics.
As a prelude to the various chemistry and safety topics addressed below, it is useful to review the types of product-related impurities that have been reported in oligonucleotide therapeutics. To this end, an attempt was made to catalog oligonucleotide impurities reported in the literature through December 2015 and to group them into broad structural types; the results of the effort are summarized in Table 1 (the interested reader is also directed to a recent excellent review of oligonucleotide impurities [10]).
SPS, solid-phase synthesis.
Chemistry Considerations
Impurity characterization
The characterization of product-related impurities is an important activity in the development of any new chemical entity. In this regard, to the extent practical, sponsors are expected to make an effort to determine the structures of all impurities present or likely to be present in significant quantities. Such an undertaking typically requires a detailed appreciation of the synthetic route and its chemical transformations, knowledge of starting materials and the fate of starting material impurities, and an understanding of potential degradation pathways. The information is used to guide the empirical process of impurity characterization itself, which may consist of a variety of steps, including impurity isolation or enrichment, chemical and physicochemical evaluation, and structural confirmation through unambiguous synthesis. Often, complete structural elucidation of impurities is one of the most challenging aspects of drug development.
Attempts should be made to characterize product-related impurities of oligonucleotide therapeutics. As discussed above, any such effort should be guided by detailed assessments of the synthetic process, starting material impurities, and potential degradation pathways. Given the anticipated challenges associated with empirical structural elucidation, the importance of the initial assessment phase cannot be overstated. However, it is also probable that the platform nature of oligonucleotide manufacturing, the ability to make straightforward predictions about the impact of starting material impurities, and the availability of a significant body of literature describing nucleic acid impurities (see, eg, Ref. [10]) combine to render the assessment phase more informative than typical for a small-molecule drug; in short, the old adage a day in the library saves a month in the laboratory appears to be especially relevant to oligonucleotide impurities.
It is also likely that the extent of platform information available for an impurity class or type will to a large extent dictate the amount of compound-specific empirical data required to characterize the same impurity class in other related oligonucleotides. In this regard, we imagine a spectrum of empirical data requirements, beginning at one end for impurities previously characterized independently by several groups for several oligonucleotides and ending at the other extreme for an impurity being reported for the first time. Impurities in the former category may include the phosphate diester impurity of phosphorothioate oligonucleotides [11], n − 1 impurities [28–30], n + 1 impurities [31], and abasic oligonucleotides [16]. In these and other similar cases of commonly observed impurities with well-understood mechanisms of formation, compound-specific empirical data requirements are likely to be minimal. For example, in the case of 2′-deoxyphosphorothioate oligonucleotides, the observation of a signal of mass 16 Da less than that of the parent oligonucleotide is probably sufficient empirical evidence of the phosphate diester impurity.
The situation is clearly different for impurities that are being reported for the first time; here a simple mass measurement, for example, although potentially useful, is by itself unlikely to constitute sufficient structural proof. This view is of course reflected in published reports describing oligonucleotide impurities, where invariably an array of experimental evidence, including impurity isolation, high-resolution mass spectrometry, NMR spectroscopy, chemical and enzymatic digestions, reactions of model systems, and unambiguous synthesis is presented in support of any structural assignment (see, eg, Refs. [21,24,27]).
The technical difficulties associated with characterizing impurities of even small-molecule drugs were alluded to. In the present case, the challenge is exacerbated not only by the relatively large molecular weights of oligonucleotide impurities (typically several thousands of daltons) but also by the fact that many exist as mixtures of closely related components. For example, the phosphate diester impurity of a 20-mer phosphorothioate oligonucleotide is a mixture of up to 19 different components that are distinguished from each other only by the location of the phosphate diester linkage. The same situation applies, for example, to the CNET impurity (impurity in which a single thymine is replaced by N3-(2-cyanoethyl) thymine [20]) of oligonucleotides that contain more than one thymine residue and the abasic impurity of molecules that contain more than a single 2′-deoxypurine residue. For composite impurities of this type, questions regarding the need to characterize the impurity class to the level of the individual components may arise. In general, such efforts should not be required and in most cases, sound chemical arguments can be made to support the composite nature of the impurity.
In summary, we suggest that an attempt should be made to characterize product-related impurities in oligonucleotide therapeutics. The extent of compound-specific empirical data required depends largely on the impurity in question; commonly observed impurities that are well described in the literature require significantly less compound-specific characterization data than impurities being reported for the first time. Finally, the technical difficulties associated with empirical characterization of oligonucleotide impurities should not be overlooked; therefore, sponsors should keep in mind that it may not be practical to determine the structures of all individual impurities. When this situation occurs, we recommend presenting a summary of the work undertaken and referring to the unidentified impurity by some suitable analytical property, such as relative retention time or mass.
Analytical methods
To the fullest extent practical, it is recommended that an effort be made to develop and deploy analytical methods capable of resolving impurities from the parent oligonucleotide and from each other. Commonly used techniques include capillary gel electrophoresis (CGE) [37,38] and anion exchange high-performance liquid chromatography[11,39,40]. More recently, ion-pair high-performance liquid chromatography (IP-HPLC) techniques have become popular and, in many instances, have replaced CGE as the method of choice for achieving length-based separations. The ability of electrospray ionization mass spectrometry (ESI-MS) to produce ions directly from a flowing solution makes it ideally suited to chromatographic hyphenation and, in recent years, the use of IP-HPLC-ESI-MS techniques has become more widespread [33,41–46]. In the case of double-stranded oligonucleotides, in addition to analyses conducted on the individual single strands, it is often important to conduct impurity tests under nondenaturing conditions, with a view to separating and quantifying residual single strands and aggregated components from the parent duplex. Important techniques for this purpose include size exclusion chromatography, ion exchange chromatography [47], IP chromatography [48,49], and capillary electrophoresis [50]. The topic of oligonucleotide analysis is the subject of two recently published books [51,52].
The variety of oligonucleotide structures in development makes it difficult to recommend any one analytical technique over another. It is also likely that additional techniques will continue to be developed. The degree to which practical resolution of impurities from the parent sequence and from each other will no doubt vary and the task will likely become progressively more difficult as the length of the parent oligonucleotide increases. However, we suggest that the goal of impurity profile testing should be to provide as complete and accurate a description of the oligonucleotide content of the sample as possible.
Impurity reporting
For small-molecule therapeutics, listing of some number of impurities in the drug substance specification is standard practice; these impurities are normally referred to as specified impurities. Significant quantities of any additional impurities not listed in the specification are also expected to be reported; these impurities are normally referred to as unspecified impurities.
Specifications for oligonucleotide therapeutics will likewise typically include tests of both specified and unspecified impurities. The former usually comprises frequently observed impurities that are diagnostic of the success or otherwise of a particular step or control strategy in the manufacturing process. For example, single-stranded oligonucleotides typically contain low levels of n − 1 impurity, the levels of which are largely dependent on successful completion of the detritylation step of solid-phase oligonucleotide synthesis. Therefore, monitoring n − 1 in the drug substance may confirm that the detritylation step performed as expected. Consequently, specifications for many single-stranded oligonucleotides will include a test of n − 1. In addition to specified and unspecified impurities associated with the manufacture of the intermediate single strands, duplex drug substance may contain excess single strands. Consequently, single-stranded intermediates are often included as specified impurities of double-stranded oligonucleotide drug substances (the impurity profiles of the single strands typically remain unchanged in the duplex, which leaves excess single strands as the only impurities to be controlled in the duplex). Similarly, monitoring commonly observed starting material-derived impurities in the drug substance may provide assurance of an effective starting material control strategy. 3
For drug product specifications, the focus will likely shift from impurities formed due to side reactions of the drug substance manufacturing process and starting material-derived impurities to degradation products and potentially impurities formed during the drug product manufacturing process. It is recommended that the selection of specified degradation products be based primarily on the results of forced degradation testing (for a recent review, see Ref. [25]; see also Ref. [19]).
The challenges associated with separating product-related impurities from the parent oligonucleotide and from each other were discussed above. For most if not all oligonucleotides, complete separation of impurities from each other will likely not be practical. Inevitably then, at least at some level, it will be necessary to report specified impurities as groups, or classes. Several means of assembling impurities into groups can be imagined. One appealing strategy is to group impurities on the basis of structural class, into categories such as n − 1, n + 1, abasic oligonucleotides, and CNET. An obvious benefit of this approach is that all members of a particular group, for example, the components that comprise n − 1, are subject to the same mechanism of formation. Therefore, grouping impurities on the basis of structure is consistent with the notion that the reported results should reflect the success or otherwise of a particular synthetic step or control strategy (in the case of n − 1, the success of the detritylation step of oligonucleotide synthesis). It is acknowledged that even this simplified approach is difficult to implement for all impurities. In these instances, grouping impurities on the basis of some analytical property such as retention time may be more appropriate.
We conclude the section on impurity reporting with a brief discussion of the reporting threshold, that is, the level above which there is an expectation to report (and sum) impurities. For most small-molecule drugs, the reporting threshold is set to 0.05% (ICH Q3A and Q3B); there is currently no generally accepted reporting threshold for oligonucleotide therapeutics. The most important consideration when establishing a reporting threshold is the lower limit of quantification (LOQ) of the method used to measure impurities; in short, it is understood that little useful information is obtained by reporting impurities that are present below the LOQ. For small-molecule drugs, where impurities are typically single components that are readily separated chromatographically and appear as sharp, well-defined peaks detected by UV absorbance, LOQ values on the order of 0.02% or less are readily achieved; it was largely by considering what is technically feasible for most small-molecule drugs that the 0.05% reporting threshold was established.
As discussed above, product-related impurities of synthetic oligonucleotides are often difficult to separate chromatographically and in many instances can only be detected using mass spectrometry. Furthermore, even when chromatographic separation is practical, the composite nature of most oligonucleotide impurities and, in the case of phosphorothioate oligonucleotides at least, their diastereoisomeric nature contribute to chromatographic peak broadening. Peak broadening, which reduces signal-to-noise, and the use of mass spectrometry detection, which has been reported to exhibit LOQ values of around 0.1% to 0.3% for oligonucleotide impurities [33,46], indicate that a higher reporting threshold will be required for oligonucleotide drugs.
While the actual value will depend on the size and complexity of the molecule in question, we cautiously suggest that a reporting threshold of 0.2% should be attainable in the majority of cases. 4 , 5
Specification limits
By definition, each specified impurity listed in the specification is associated with a specification limit, and comparison of drug substance and product results against specification limits is an important part of determining the suitability of the material under test.
In the case of product-related oligonucleotide impurities formed due to side reactions of the manufacturing process, it will likely be appropriate to develop limits on the basis of data obtained from analysis of drug substance and drug product batches made by processes that resemble closely the intended commercial processes. To ensure that specification limits are consistent with reasonable manufacturing variability, it will also be important to develop an understanding of the impact of various process parameters (inputs) on such by-products. Process knowledge of this type is normally gathered during the development phase on an appropriately scaled model system. Large- and small-scale platform manufacturing experience, that is, experience gained in the synthesis of related oligonucleotides, might also be considered. Specification limits for oligonucleotide degradation products are probably best derived by consideration of accelerated stability data. All limits should also be consistent with analytical method capabilities and the available safety information (vide infra).
The considerations outlined above for the development of limits applicable to by-products and degradation products of oligonucleotides are no different from those made for small-molecule drugs. However, the arguments made in support of starting material-derived impurity limits for oligonucleotides may be unique to oligonucleotides. 6 This is primarily because levels of starting material-derived impurities in oligonucleotides are generally unresponsive to changes in process parameters. In fact, experience suggests that levels of starting material-derived impurities are solely determined by (critical) impurity levels in starting materials and by starting material incorporation frequencies. This is in contrast to many small-molecule processes, where downstream processing and purification steps often tolerate relatively large variations in starting material input. Consequently, the ability to control starting material-derived impurities depends solely on the ability to control reactive impurities in starting materials. This means that in contrast to product-related impurities formed due to side reactions, where drug substance batch history data can be used to help justify specification limits, batch history data are of limited value when developing suitable limits for starting material-derived impurities. The situation is exacerbated by the linear nature of solid-phase oligonucleotide synthesis, which guarantees that even small quantities of reactive starting material impurities result in significant quantities of drug substance impurities. Therefore, we recommend that limits for starting material-derived impurities be developed primarily on the basis of the control strategies in place for starting materials and that less importance be attached to drug substance batch data. Of course, regardless of the justification approach used, limits for starting material-derived impurities should be consistent with available safety information.
Safety Considerations
Safety considerations are centered on the notion of impurity qualification, which is defined for small-molecule drugs as the “process of acquiring and evaluating data that establish the biological safety of an individual impurity or a given impurity profile at the level(s) specified” (ICH Q3A and Q3B). The ideas developed in Q3 are predicated on the fundamental principle that patients ought not to be exposed to impurity amounts above those for which safety has either been demonstrated or, by consideration of a variety of factors, reasonably assumed. This in turn leads to the general requirement that specification limits for specified impurities set above the qualification threshold be below those deemed safe (Fig. 1).
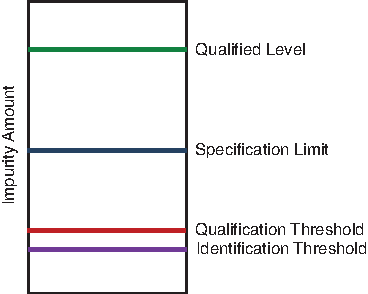
Relationship between safety data (qualified level), specification limits, and identification and qualification thresholds. 7
This same underlying principle generally applies to oligonucleotide therapeutics and, in what follows, we have tried to provide practical scientific advice on the process of qualifying product-related oligonucleotide impurities.
Initial questions with regard to qualification include impurity thresholds, specifically the level above which there may be a requirement to assess safety, the structure of the impurity, and the nature of the data required to make the assessment. We propose to leave aside for now the consideration of impurity thresholds, and consider first data-type requirements (we return to the first question in the discussion of identification and qualification thresholds). It is proposed that data-type requirements be dictated primarily by impurity structure. Specifically, for the purposes of qualification, we suggest that impurities should be assigned to one of four distinct classes on the basis of their structure, and that each class be associated with particular data-type requirements (Table 2).
Assumes that at the specification limit, the individual components of the impurity are each present below the qualification threshold.
Safety assessment required if specification limit is higher than the qualification threshold.
Class I comprises impurities that are also major metabolites of the parent oligonucleotide. Examples include impurities that are structurally related to the parent by loss of nucleotides from one end or the other. Because they are likely to be structurally identical to the major products of exo- and endonuclease-mediated metabolism [53], such impurities are qualified during the nonclinical safety assessment and should require no additional safety testing. A second example is the case of a conjugated oligonucleotide designed to liberate the corresponding unconjugated molecule in vivo; here there ought to be no need to assess the safety of the unconjugated oligonucleotide (which may be listed as a specified impurity of the conjugated molecule). Because they are likely to be present in vivo, it also seems appropriate to expand the metabolite class to (parent) single-stranded impurities of double-stranded drugs.
Class II impurities contain only structural elements found in naturally occurring nucleic acids. Examples include the phosphate diester impurity of phosphorothioate oligonucleotides and impurities of RNA that arise from migration of internucleotide linkages. The fact that these structural elements occur endogenously rules out inherent safety concerns associated with their presence in oligonucleotide drugs. It should be noted that we choose not to include in this class impurities that contain structural elements that arise in vivo through damage of endogenous nucleic acids. For example, we chose not to regard the abasic site as a structural element of endogenous nucleic acid as part of this class despite the fact that it accounts for a large fraction of the damage suffered by DNA. This decision, which is extended to other products arising from DNA damage such as oxidized bases and photodegradation products, recognizes that such lesions are often repaired rapidly and that, as a consequence, conclusions regarding the safety of the same structural elements in oligonucleotide drugs, which are presumably not subject to repair processes, are more difficult to make.
Class III impurities comprise those that differ from the parent oligonucleotide on the basis of sequence, but are otherwise not distinguished by the presence of new structural elements. Common examples include n − 1 and n + 1 impurities (Table 1), but others are also possible. For example, degradation products caused by deamination, where uracil (or thymine) substitutes for cytosine (or 5-methylctosine) at one position [25]. In the absence of structural elements not found in the parent oligonucleotide, any potential toxicities associated with impurities of this class are likely restricted to those arising from off-target antisense effects or from creation, by nucleotide removal (n − 1), insertion (n + 1), or substitution (eg, deamination), of an inflammatory sequence motif.
Although these impurities could possibly hybridize with other RNA targets, levels of the individual components that comprise n − 1, n + 1, and other sequence impurities are generally far too low to have any pharmacologic effect. For example, in the case of the n − 1 impurity, which is normally the most abundant of the common sequence impurities, levels of the individual components typically do not exceed a few 10ths of a percent of the parent oligonucleotide.
As alluded to above, in addition to potential antisense-mediated toxicity, it is also possible that deletion, insertion, or substitution of a nucleotide may create an inflammatory sequence motif. For example, deletion of the central nucleotide from an oligonucleotide of sequence “…CpTpG…” creates an n − 1 impurity that contains the CpG dimer [54]. Here again, however, it is very likely that the quantities of such impurities will be too low to impact the safety profile of the parent molecule. 8
With regard to the safety of sequence impurities therefore, because their potential toxicities are predicated on the sequences of the individual components, it makes sense to consider the components separately. This is in contrast to impurities that possess structural elements not found in the parent oligonucleotide or in naturally occurring nucleic acids, where safety assessment of the sum of the individual components may be more appropriate. Therefore, because the individual components of impurities such as n − 1 and n + 1 are present at levels that almost certainly do not approach the proposed qualification threshold (see Identification and Qualification Thresholds section), we believe in general that animal qualification studies should not be necessary.
Class IV impurities contain structural elements not found in the parent oligonucleotide or in naturally occurring nucleic acids, and impurities of unknown structure. In instances where specification limits in excess of the qualification threshold are desired, we recommend the safety of the impurity be evaluated in nonclinical toxicity studies (vide infra).
Qualification strategies
For those impurities that have structural elements not found in either the parent oligonucleotide or in naturally occurring nucleic acids (Class IV), the qualification approach follows the general principles outlined for traditional small molecules. The most direct means of qualifying impurities early in development is to perform the initial toxicity studies with the same batch of material that will be used for the initial clinical studies. The margins between the clinical doses studied and any test article-related nonclinical effects generally cover impurity levels. Alternatively, initial toxicity studies may be performed with a batch manufactured by the same process as that intended for clinical material, although modified deliberately to increase impurity levels. For example, wider pooling criteria could be used during purification of the toxicology batch than of clinical batches. In later development, data from later phase toxicity studies using batches with different impurity contents can be used to expand coverage levels for impurities that were not covered by existing toxicity studies. However, at any phase during clinical development or indeed subsequently, a batch may be produced that contains either (1) a novel impurity in an amount that exceeds the qualification (or identification) threshold or (2) a known impurity in an amount that exceeds the available toxicology coverage. These scenarios may necessitate toxicological bridging studies.
A standard approach to qualification of an impurity that is not (sufficiently) covered by available nonclinical or clinical data is to conduct a dedicated nonclinical safety study. Although the details of such studies will likely depend on the nature of the drug and the impurities in question, the authors offer the following general advice.
Species, test article design, study duration, and dose levels
Consistent with the advice provided in Q3A, it is recommended that studies designed for the purpose of qualifying oligonucleotide impurities should be conducted in a single rodent species. With regard to test article design, there are in principle two main approaches. First, one may choose to manufacture test articles that comprise solely the impurity or a mixture of impurities. However, it is preferred to use test articles comprising parent oligonucleotide fortified with the impurity or impurities of interest. There are several reasons for this preference, among which is that it is often more practical synthetically to produce oligonucleotides that are enriched in an impurity than it is to produce the impurity in its pure form. Of course, and perhaps more importantly, assessing the safety of the impurity in the presence of the parent oligonucleotide is more representative of the true conditions under which the impurity is presented to the patient.
Many common oligonucleotide impurities, for example, abasic and CNET impurities, are mixtures of individual components that differ from each other only in the location of the lesion within the sequence. Consequently, one may enquire whether it is necessary to assess the toxicity of all possible members of a given impurity class, or whether assessment of a single representative example is sufficient. In our opinion, assessment of a single representative impurity of each class will normally be sufficient, but assessment of mixtures of the same impurity class is certainly permissible and, due to synthetic considerations, sometimes practically unavoidable (the single-impurity-sufficiency position is also logically consistent with the platform arguments provided below). It is also our experience that impurities are most efficiently assessed in a cassette manner, by dosing test articles comprising the parent oligonucleotide fortified with more than one class of oligonucleotide impurity.
In general, study duration should be sufficient to ensure that steady-state concentrations of drug are achieved in the tissues of interest. In the case of phosphorothioate oligonucleotides that display relatively long half-lives [53] and that are designed to be taken chronically, durations of up to 13 weeks may be required. For drugs that exhibit shorter tissue half-lives or drugs not intended for chronic administration, shorter studies may be sufficient. Dosing will depend to some degree on test article design and the level of coverage desired, but clearly the amount of impurity dosed should equal or exceed the level for which coverage is sought. It is also recommended that animals be dosed at a minimum of two levels, the lower of which should be that expected to emerge as the NOAEL, whereas the higher dose should expand the therapeutic window or be sufficient to produce toxicological findings consistent with those of the parent oligonucleotide (see below for a rationale). In addition to groups dosed with impurity-enriched test articles, control groups dosed with the parent oligonucleotide and with vehicle only should be included. A sufficient number of animals, for example, 10 of each gender, should be included in each arm of the study.
Toxicological assessment
Toxicological assessments should include in-life observations such as body weight, food consumption, clinical observations, and ophthalmic examinations; clinical pathology, including hematology and clinical chemistry, and postmortem evaluations such as gross necropsy, organ weights, and histopathological evaluations. Tissue concentrations of the parent oligonucleotide should be measured to document exposure.
Calculating toxicology coverage
There are perhaps two main ways in which the toxicity data generated in the study can be used to calculate coverage. A simple way is to express the impurity content of the test article in units of percent and to set impurity coverage equal to this value. This approach is valid provided the safe dose level determined in the toxicity study equals or exceeds the clinical dose. An alternative, more flexible approach is to calculate the actual amount of impurity present in the test article at the appropriate dose and to use this value to calculate coverage, again expressed as a percent of the clinical dose. These alternate approaches are illustrated by example in Table 3. 9
In units of mg total oligonucleotide dosed/kg/week.
In units of mg parent oligonucleotide dosed/kg/week.
Equal to: impurity level in test article (5%) × highest safe dose level (10 mg/kg/week).
Equal to: amount of impurity demonstrated safe (0.5 mg/kg/week)/clinical dose (5 mg/kg/week) × 100%.
When defining the highest safe dose level of an oligonucleotide impurity, there may be instances where toxicity information obtained at dose levels in excess of the NOAEL could be leveraged. The authors believe that such arguments can be made in instances where the toxicological properties of the parent oligonucleotide are well understood and where fortification of the parent oligonucleotide with impurities does not change the extent or character of the observed toxicities. For example, if the types and severities of toxicities observed are the same at a comparable dose for the parent compound and the impurity-enriched mixture, and no new toxicities are identified, then a reasonable conclusion would be that the impurity is not contributing to the observed toxicity. In these circumstances, the toxicology findings observed are likely due to the parent oligonucleotide rather than the added impurities and, therefore, an argument can be made that the amounts of impurities present at these dose levels are qualified.
As a means of illustrating some of the above points, an impurity qualification study conducted by one of the authors (S.H.) is provided below. The study was conducted for a phosphorothioate oligonucleotide in adult CD-1 mice to evaluate systemic effects of the parent oligonucleotide enriched with a variety of impurities. Three separate impurity-enriched test articles were prepared to accommodate 10 different impurities. Each of the impurities was synthesized as a mixture of components of the same structural class. The test articles were dosed at 5 and 25 mg/kg/week of total oligonucleotide with fractionally higher impurity content compared to the parent oligonucleotide for 13 weeks. Additional groups of animals were dosed for the same duration with the parent oligonucleotide alone at 5 and 25 mg/kg/week, or with saline. Each group comprised six male and six female mice. The experimental design is summarized in Table 4.
In units of mg total oligonucleotide dosed/kg/week.
Equal to: dose × %Content.
2′-O-butyl, 2′-O-(2-ethoxyethyl), 2′-O-CH3, impurity in which a single 2′-O-(2-methoxyethyl) nucleotide (2′-MOE nucleotide) is replaced by the corresponding 2′-O-Butyl, 2′-O-(2-Ethoxyethyl), or 2′-O-CH3 nucleotide; ADP, IDP, impurity in which a single guanine is replaced by N2-acetyl-2,6-diaminopurine and N2-isobutyryl-2,6-diaminopurine, respectively [21]; CNET, impurity in which a single thymine is replaced by N3-(2-cyaoethyl)thymine [20]; Abasic impurity, impurity that contains a single abasic site; Dithioate, impurity in which a single phosphorothioate diester group is replaced by a phosphorodithioate diester group; inverted isomer, impurity that contains a single nucleotide inserted to make a 3′-3′ and a 5′-5′ phosphorothioate diester linkage; 3′-MOE, impurity in which a single 2′-MOE nucleotide is replaced by the corresponding 3′-MOE nucleotide.
Toxicological assessments included in-life observations (body weight, food consumption, clinical observations, ophthalmic examinations), clinical pathology (hematology and clinical chemistry), and postmortem evaluations (gross necropsy, organ weights, histopathological evaluations). Tissue concentrations of the parent oligonucleotide were measured from liver samples taken to document exposure.
The parent oligonucleotide and the three impurity mixtures were well tolerated at 5 and 25 mg/kg/week. Test article had no effects on morbidity, mortality, clinical findings, body weights, food consumption, ophthalmic examinations, hematology, gross findings at necropsy, or organ weights. No meaningful differences were apparent among treatment groups for clinical chemistry parameters at any dose level. Occasional individual animals had mild increases in alanine aminotransferase (ALT). These findings may have been related to the microscopic findings in the liver (eg, vacuolated/granular macrophages), which were considered test article related. Mice in all test article groups (parent oligonucleotide and impurity mixtures) had microscopic evidence of vacuolated/granular macrophages in tissues throughout the body and accumulation of basophilic granules within the tubular epithelium of the kidneys. The microscopic findings were consistent with oligonucleotide uptake and/or cellular activation and cytokine production due to proinflammatory effects of oligonucleotides and were not considered adverse. No additional microscopic findings were observed in animals treated with the impurity mixtures.
In summary, enriching the parent oligonucleotide with the indicated amounts of impurities did not change the NOAEL in mice or the character of the common class effects observed in animals dosed with the parent oligonucleotide alone. The results of the study indicate impurities qualified in the amounts listed in Table 5.
In the authors' opinion, the results obtained in the above study and other studies of similar design are sufficient to qualify the same impurity classes to the amounts indicated in related oligonucleotides. The concept of a sequence-independent oligonucleotide impurity toxicology, which is consistent with the idea that the oligonucleotide sequence surrounding a modified internucleotide linkage, sugar, or base is unlikely to impact the physicochemical properties of the lesion significantly, is aligned with database structural alert approaches applied to small molecules (eg, ToxAlerts, DEREK, NOXNET [55]), and means that animal studies should not need to be repeated to qualify the same impurity classes in other oligonucleotides of the same chemical structure.
Identification and Qualification Thresholds
Another important question that applies to impurity qualification is at what level, or in what amount, does an impurity need to be present to warrant being considered for qualification? We now turn to the important topic of establishing identification and qualification thresholds for oligonucleotide impurities.
The concept of dose-variable identification and qualification thresholds is discussed in ICH Q3A and Q3B. At doses of up to 2 g/day, the identification and qualification thresholds for small-molecule drug substance and drug product impurities are 0.10%, and 0.15% or 1.0 mg, respectively (in each case, the smaller of the two quantities is applied). The identification threshold establishes the level above which there is a requirement to qualify impurities of unknown structure, and it is standard practice in drug substance and drug product specifications to set the limit on unspecified impurities equal to this value. The qualification threshold establishes the level above which there is a requirement to qualify impurities of known structure that do not contain structural elements known to present a hazard to human health (impurities that contain such structural elements, eg, genotoxic impurities, require separate consideration). Consequently, the qualification threshold mandates that all specification limits for specified impurities set higher than the qualification threshold be supported by qualification data.
The identification threshold is driven by quality (chemistry) expectations. Specifically, the general quality expectation is that the formation of new and unidentified impurities above some limit, that is, the identification limit, be avoided. Implicit in this expectation is that manufacturing processes, control strategies, and analytical methods are understood and developed to the extent that they are capable, when operating in a state of control, of rendering drug substance or drug product that contains less than identification threshold levels of new impurities; for most small-molecule drugs, this value is 0.10%.
A generally accepted identification threshold has not been established for oligonucleotide impurities, although it would clearly be advantageous to do so. Interestingly, progress has been made toward establishing thresholds for synthetic peptide drugs and, since 2009, European Pharmacopeia (Ph. Eur) monograph 2034 has included identification and qualification thresholds of 0.5% and 1.0%, respectively, for peptides produced by chemical synthesis.
The Ph. Eur. identification threshold for peptides is considerably higher than that established for small molecules. This is the case for at least two reasons. First, most peptides are produced by solid-phase synthesis (SPS) or by a combination of solid-phase and solution-phase synthesis. Although highly efficient, the nature of SPS, where the products of one reaction are fed into the next without purification, presents very little opportunity for impurity removal. Consequently, the presence of even minute quantities of reactive impurities in starting materials, for example, may lead to significant quantities of drug substance impurities. Second, the iterative nature of SPS and the complex nature of peptide drugs create rather complex impurity profiles, where it is often difficult analytically to distinguish impurities from the parent peptide and each other. These factors conspire to place practical limitations on what might reasonably be achieved in the control of unspecified impurities.
Of course, because oligonucleotides are also made by SPS, the manufacturing considerations described above that led to the adoption of a higher identification threshold for peptides apply equally to oligonucleotide therapeutics. Therefore, the authors believe that the identification threshold for oligonucleotide drugs also need to be set higher than that established for small molecules [this position is echoed in recent draft guidance published by PMDA (Considerations and Challenges in the Development of Nucleic Acid Drugs–Interim Report, March 31, 2015)]. We suggest an identification threshold of 1.0%, which we contend is about the minimum level to which it will be possible to control unspecified impurities in most oligonucleotide therapeutics. 10
Having argued the appropriateness of a 1.0% identification threshold from the quality (or chemistry) perspective, we must now ask whether such a value can be considered suitable from the safety perspective. We believe there are several reasons for answering in the affirmative. First, the nature of oligonucleotide synthesis and the potential sources of new, unspecified oligonucleotide impurities make it very likely that the latter will be structurally closely related to the parent oligonucleotide. Specifically, unspecified oligonucleotide impurities will, like most impurities identified to date, comprise the parent oligonucleotide modified at a single internucleotide linkage, sugar, or heterocycle. Because they are modified at only a single site, the physicochemical properties of unspecified impurities are likely to be very similar to those of the parent oligonucleotides. That is, the impurities will be water soluble polyanions of similar molecular weight and protein binding properties to the parent oligonucleotide. Because these are the properties that influence class-related tolerability and PK/metabolism of oligonucleotide drugs, the toxicities of new, unspecified oligonucleotide impurities are expected to be similar to those of all impurities tested to date and indistinguishable from those of the parent oligonucleotide.
Second, because of the large difference in molecular weights between oligonucleotide and small-molecule impurities, the same quantities of each represent very different molar amounts. For example, comparison of a typical small-molecule drug impurity of molecular weight = 500 Da to an oligonucleotide impurity of molecular weight = 5,000 Da reveals a 10-fold difference in molar amounts for the same quantity of material. Therefore, at the same clinical dose, the molecular weight difference suggests that controlling an unspecified oligonucleotide impurity to 1.0% is approximately equivalent, in terms of molar amounts, to controlling unspecified impurities in a typical small-molecule drug substance to 0.10%. Third are the available toxicology data for qualifying common impurities of oligonucleotides, including data from the types of dedicated qualification studies described above. In regard to these data, the authors are not aware of any toxicity attributable to an impurity.
Having established the appropriateness of a 1.0% identification threshold, which by definition applies to unspecified impurities about which no empirical structure information may be available, it seems consistent to propose a qualification threshold in excess of this value. The implicit assumption here is that determining the structure of an impurity, and confirming the absence of features known to present a hazard to human health, provides an additional degree of safety assurance missing in the case of unidentified impurities. For small-molecule impurities, the qualification threshold is set 50% higher than the identification threshold; we therefore suggest a qualification threshold for identified oligonucleotide impurities of 1.5%.
Conclusions and Future Work
In this white paper, we provide scientific advice on a variety of chemistry issues concerning oligonucleotide impurities. Despite their chemical complexity, it is our experience that with sufficient effort and imagination, most oligonucleotide impurities can be characterized with at least some degree of precision, if not with atomic resolution. It is also our experience that characterization efforts often lead to process improvements, which ultimately lead to higher purity drugs. We believe strongly that similarities among oligonucleotide drugs and their methods of synthesis provide an almost unparalleled opportunity to leverage chemical information obtained from the study of one molecule to another; it is in the spirit of this platform approach that we encourage and applaud all efforts to publish work describing oligonucleotide impurity characterization.
This article also discusses the important topic of impurity qualification. We provide a holistic approach that differentiates qualification requirements on the basis of structure and level; the approach is summarized in decision tree format in Fig. 2. In common with our views on chemistry matters, we believe that structural similarities among oligonucleotide therapeutics provide an important opportunity to share safety information, most profitably in the area of animal impurity qualification data such as those summarized in Table 5. Therefore, we encourage publication of such studies in the firm belief that doing so will inform and reduce the risk to patients treated with oligonucleotide therapeutics, while at the same time make it unnecessary to evaluate in animals impurities for which safety has already been established.
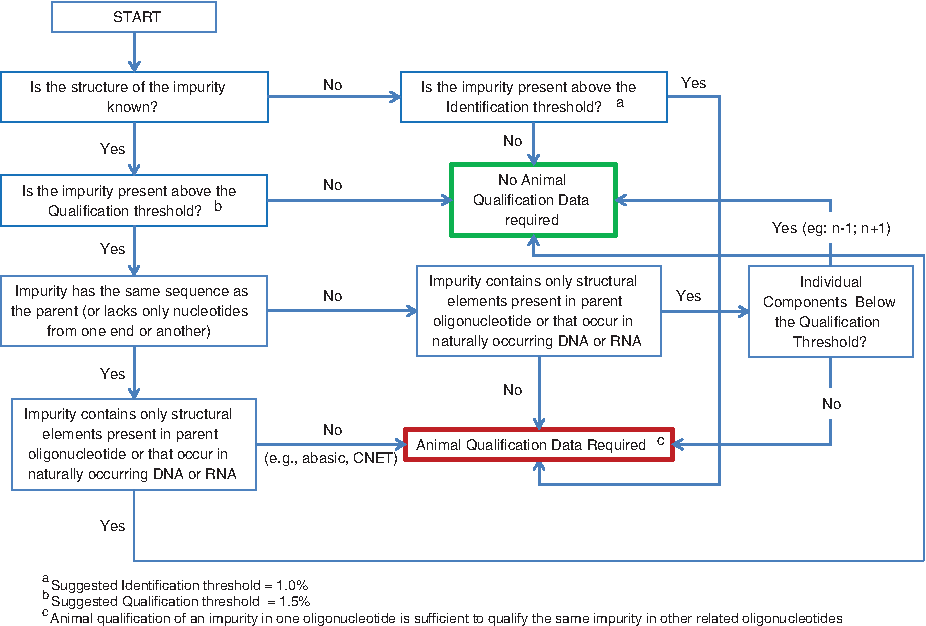
Decision tree for oligonucleotide impurity qualification.
Footnotes
Acknowledgments
The authors thank Dr. Rene Thürmer (BfArM) and Dr. Gunther Boos (BfArM) for useful discussions.
Author Disclosure Statement
D.C. and S.H. are employed by Ionis Pharmaceuticals, Inc.; A.T. and N.A. are employed by AstraZeneca; C.d.B. is employed by ProQR Therapeutics; S.G.-S. and M.K. are employed by Alnylam Pharmaceuticals, Inc.; N.S. is employed by Anavex Life Sciences; B.A. is employed by GlaxoSmithKline; B.B. is employed by ISA Therapeutics B.V.; and J.F. is employed by Celgene. No competing financial interests exist.