
Abstract
Recently, some studies have reported nephrotoxicity associated with a certain class of antisense oligonucleotides (ASOs) in humans. One possibility for reducing the potential nephrotoxicity of ASOs is to alter their pharmacokinetics. In this study, we investigated the effect of a ligand conjugation strategy on the renal accumulation of ASOs. We selected two ligands, cholesterol and N-acetylgalactosamine (GalNAc), with the purpose of reducing renal distribution and liver targeting, and then designed a series of cholesterol–GalNAc dual conjugated ASOs. The gene-silencing activity of the cholesterol-GalNAc dual conjugated ASO in the liver was slightly lower than that of a GalNAc-conjugated ASO. On the other hand, the renal distribution of the cholesterol–GalNAc dual conjugated ASO was considerably decreased compared with the GalNAc-conjugated ASO, as we expected. As dual conjugation was successful in reducing the renal distribution of ASO, it should be an effective strategy for reducing the nephrotoxic potential of ASOs.
Introduction
A
However, as clinical experience with ASOs has increased, some safety concerns have been raised [8,9]. Although uncommon, one of the adverse events reported is nephrotoxicity that appears to be related to ASO chemistry and renal drug burden [9]. A noteworthy precedent is that serious acute kidney injury was found in healthy subjects during the clinical development of SPC5001, a locked nucleic acid (LNA)-modified ASO for the treatment of dyslipidemia, and the trial was eventually terminated in spite of the agent's significant therapeutic activity [10,11]. More recently, marked elevation of serum creatinine and the urinary kidney injury molecule 1 (Kim-1) was observed in subjects receiving a multiple-dose protocol of 2′-methoxyethyl (MOE)-modified ASO targeting sodium–glucose cotransporter-2 (SGLT2) in the first-in-human study [12,13], which had not been predicted from preclinical animal studies [14].
Our group has been independently developing ASOs targeting liver-associated diseases [15]. During preclinical development, we also observed nephrotoxicity, which will be reported elsewhere. Therefore, nephrotoxicity can be considered to be a potential adverse event of a certain class of ASO agents that needs to be eliminated. In this context, reducing the renal distribution of ASOs would be a preferred strategy because nephrotoxicity is a dose-related event [11–13]. To reduce the possibility of nephrotoxic effects of ASOs, we investigated ligands that would be capable of conjugation with ASOs and reduce their renal distribution, while maintaining the gene-silencing activity in the liver, the target organ.
Amazing advances have been made in techniques for the delivery of ASOs to the liver over the last few decades. Hepatic accumulation of ASOs has been successfully increased through phosphorothioate (PS) internucleotide modification [16,17]. PS-ASOs are thought to interact more strongly with serum proteins such as albumin [18], and they have been found to be taken up by hepatocytes mainly through Stabilin receptors [19]. In addition, cholesterol conjugation strategies have come to be widely utilized for delivering oligonucleotides to the liver [2,20,21]. It has been found that cholesterol conjugation helps oligonucleotides to interact with serum proteins and lipoproteins to trespass into hepatocytes through receptor-mediated or nonmediated pathways [20,22–24]. Furthermore, in addition to the liver–acid characteristics of cholesterol, its ability to prolong the blood half-life of cholesterol-conjugated ASOs should be beneficial in reducing the total amount of ASO presented to and accumulated in the kidneys [22]. We have recently investigated the effect of cholesterol conjugation on the in vivo gene-silencing activity of ASOs, and found the linker structure between ASO and cholesterol to be important [25]. Nakajima et al. took this a step further and investigated the optimum location for binding cholesterol to ASOs [26]. In this regard, it is important to note that Watanabe et al. showed that cholesterol-conjugated ASOs preferentially accumulated in nonparenchymal cells (NPCs) over hepatocytes in the liver [27].
On the other hand, N-acetylgalactosamine (GalNAc) has been gaining more attention as a liver-targeting ligand [27–30]. GalNAc conjugation permits hepatocyte-specific delivery of ASOs because multivalent GalNAc is a specific ligand for the asialoglycoprotein receptor (ASGPR) expressed in hepatocytes [31,32]. We and other groups have demonstrated that GalNAc improves efficacy 5–10 times as compared with the parent ASO [30,33]. Taken together, the above observations, suggest that cholesterol–GalNAc dual conjugation would enable us to reduce ASO amounts in the kidney by altering the pharmacokinetics and pharmacodynamics of ASOs.
In this study, we designed a series of cholesterol and GalNAc doubly modified hybrid ASOs against the mRNA of apoliporotein B (apoB), a prospective therapeutic target for homozygous familial hypercholesterolemia [34], and investigated their in vivo gene-silencing activity as well their effects on renal distribution.
Materials and Methods
Oligonucleotides
Monovalent GalNAc phosphoramidite was synthesized according to the procedure in our previous study [33]. The ASOs used in the present study were synthesized by Gene Design, Inc. (Osaka, Japan). (Supplementary Fig. S4–S6 Supplementary Data are available online at www.liebertpub.com/nat); The sequences of the ASOs targeting apoB were as previously reported [35] and their designs are shown in Fig. 1.
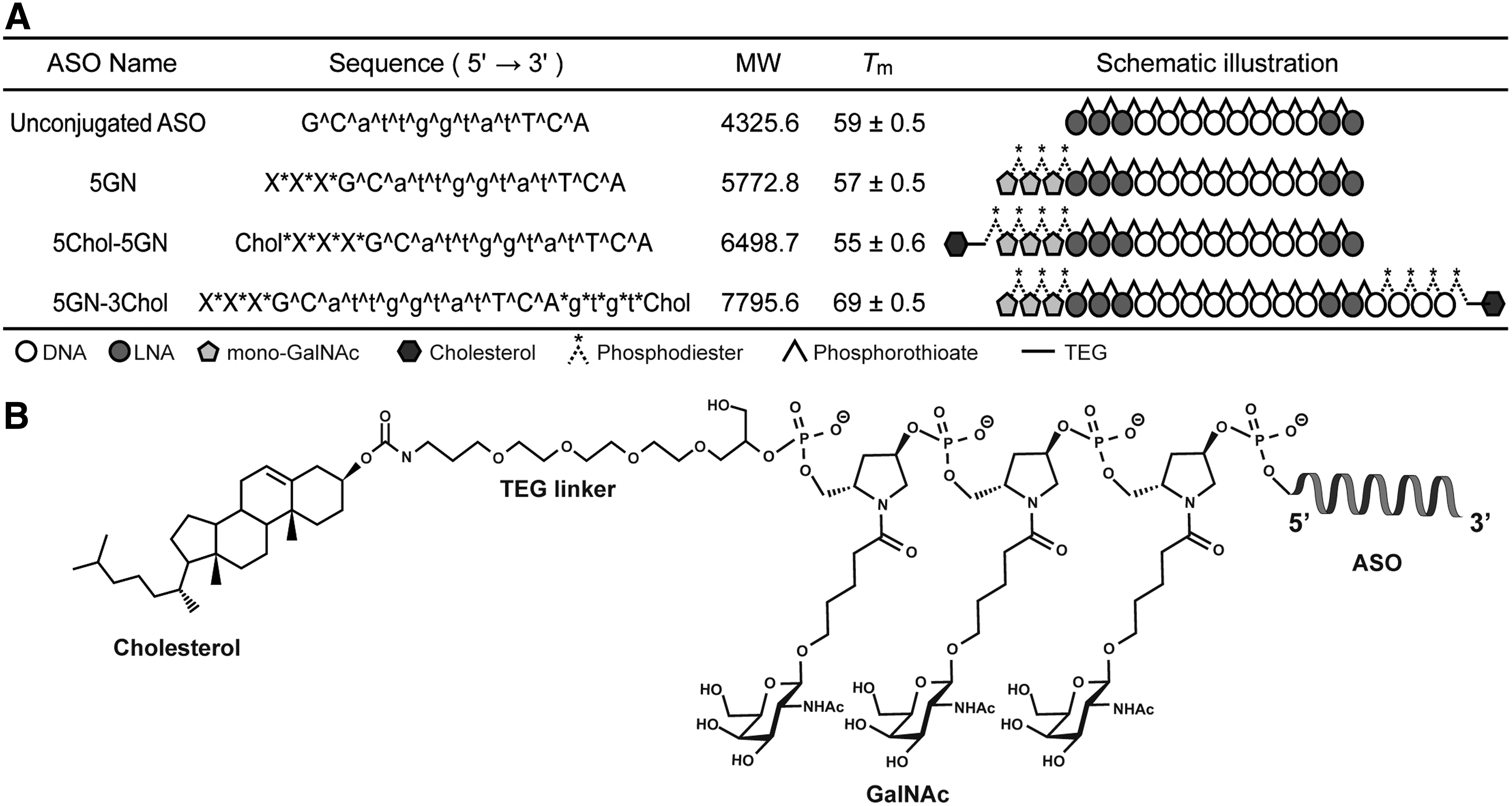
Sequences and designs of ASOs used in this study.
Tm measurement
Tm measurements were carried out on a SHIMADZU UV-1800 spectrometer with quartz cuvettes. Equimolecular amounts of each ASO and target 30-mer RNA (partial sequence of murine apoB mRNA, GenBank number NM_009693.2: 5′-gucaucacacugaauaccaaugcuggacuu-3′) were dissolved in 10 mM sodium phosphate buffer (pH = 7.2) containing 100 mM NaCl to give a final strand concentration of 2 μM. For annealing, the mixed samples were heated to 95°C, followed by slow cooling to room temperature. Melting profiles were recorded at 260 nm from 5°C to 95°C at a scan rate of 0.5°C/min. Tm values were determined from maxima of the first derivative of the melting curves.
Animal study
All animal studies were conducted with the approval of the Animal Care Ethics Committee of the National Cerebral and Cardiovascular Center Research Institute (Osaka, Japan) based on the 3Rs (Replacement/Reduction/Refinement) principle. Male C57BL/6J mice were purchased from SLC Japan (Tokyo, Japan). All studies were started when mice were 7 weeks old. Mice were maintained on a 12-h light/12-h dark cycle and fed a normal chow (CE-2; CLEA Japan, Tokyo, Japan). Each ASO was injected into mice (n = 4) intravenously at a dose of 8.75–35 nmol/kg and the injection volume was adjusted to 200 μL with saline (Otsuka Pharmaceuticals, Tokyo, Japan). Three days after ASO injections, mice were sacrificed. They were anesthetized with isoflurane (Mylan, Inc., Canonsburg, PA) before sacrifice, and whole blood samples, livers, and kidneys were collected. The livers and kidneys were flash frozen in liquid nitrogen or immersed in RNAlater (Thermo Fisher Scientific, Inc., Waltham, MA) and stored at −80°C until analysis. Serum was obtained using BD Microtainer yellow (BD, Franklin Lakes, NJ).
Quantitative polymerase chain reaction analysis
Total RNA was extracted from each tissue stored in RNAlater using the QuickGene RNA Tissue Kit SII (Fujifilm, Tokyo, Japan) with a QuichGene 810 instrument (Fujifilm). cDNA was synthesized using 2 μg of the extracted total RNA with a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Inc.). The quantitative reverse transcription–polymerase chain reaction (PCR) was performed using Fast SYBR Green Master Mix (Thermo Fisher Scientific, Inc.) with a StepOnePlus system (Applied Biosystems, Foster City, CA). Primer sequences are shown below.
Mouse Gapdh (forward: 5′-TGTGTCCGTCGTGGATCTGA-3′; reverse: 5′- TTGCTGTTGAAGTCGCAGGAG -3′).
Mouse ApoB (forward: 5′-TCCTCGGTGAGTTCAATGACTTTC-3′; reverse: 5′- TGGACCTGCTGTAGCTTGTAGGA-3′).
Serum chemistry
All parameters of serum chemistry were measured using FUJI DRI-CHEM SLIDE TCHO-PIII (Fujifilm) with a FUJI DRI-CHEM instrument (Fujifilm).
ASO quantification
The ASOs accumulated in each tissue were quantified by enzyme-linked oligonucleotide sorbent assay (ELOSA) as described previously [36]. In brief, liver or kidney homogenates containing 300 μg (liver) or 250 μg (kidney) of total protein were added to the ligation buffer (66 mM Tris–HCl, 6.6 mM MgCl2, 10 mM DTT, and 0.1 mM ATP, pH 7.6) containing a 5′-biotinylated LNA probe (5′-Biotin-TGAATA-3′) and a 3′-digoxigenin (DIG)-conjugated 5′-phosphorylated LNA probe (5′-pCCAATGC-N(6)-DIG-3′) complementary to an Apob-targeted ASO, and T4 DNA ligase (1.5 U/well; TaKaRa, Shiga, Japan). In probe sequences, upper case letters indicate LNAs (C = 5-methy-cytidine LNA) and N(C6) indicates an aminohexamethylene linker. The reaction mixture was applied to a nunc immobilizer streptavidin 96-well black plate (Thermo Fisher Scientific, Inc.) and incubated for 2 h at 15°C. After incubation, the reaction wells were washed four times with TBS-T. Anti-DIG antibody–alkaline phosphatase conjugate (Roche, Indianapolis, IN) was diluted to 1:2,000 in TBS-T containing 10% SuperBlock (TBS) Blocking Buffer (Thermo Fisher Scientific, Inc.). Then, 100 μL of the antibody solution was added to each well and incubated for 90 min at 37°C. After incubation, the reaction wells were washed four times with TBS-T. The antibody-bound ASO was detected using AttoPhos® Fluorescent AP Substrate System (Promega, Madison, WI) with CORONA GRATING MICROPLATE READER SH-9000Lab (CORONA ELECTRIC Co., Ibaraki, Japan).
Statistics
For Tm measurement, data were collected in triplicate for each ASO. In all animal studies, there were more than four mice per treated arm. Quantification of ApoB mRNA and measurement of serum chemistry were performed for the individual experiments (n = 4–5 mice). Quantification of ASO in the liver or kidney by ELOSA was performed as triplicate technical replicates for individual experiments (n = 4–5 mice). All data are indicated as mean + standard deviation. Statistical significance (P < 0.05) was determined by Tukey's multiple comparison tests or Student's t-test.
Results and Discussion
Designs of cholesterol–GalNAc dual conjugated ASOs
We utilized the previously reported monomeric GalNAc unit (mono-GalNAc) [33,37] to achieve dual conjugation. With the use of mono-GalNAc, it is possible to place the cholesterol molecule right next to the GalNAc ligand and conjugate as many GalNAc ligands as required, no matter where it is intended to place them. This is unlikely to be achieved with the conventional trivalent GalNAc [29,30]. Using mono-GalNAc, we designed and synthesized ASOs targeting apolipoprotein B (apoB) (Fig. 1).
As liberation of both cholesterol and GalNAc from the parent ASO within the target tissue or cells is considered the key to achieving high activity [25,26,33], we used a biolabile phosphodiester linkage for the cholesterol–GalNAc, GalNAc–ASO, and cholesterol–ASO linkages. As it had been found that direct conjugation of the conventional GalNAc to the 3′-end of the ASO slowed the metabolic release of ASO from the conjugate as compared with the 5′-end [38], we thought that it might be much the same for cholesterol conjugation [26]. Therefore, in the
In vivo gene-silencing activity and accumulation of cholesterol–GalNAc dual conjugated ASOs in liver
To optimize the position of the cholesterol in the parent GalNAc-conjugated ASO,

Effects of ASOs on liver and serum total cholesterol levels.
This is supported by the finding that GalNAc-conjugated ASO accumulated and showed gene-silencing activity highly specifically in hepatic parenchymal cells as compared with unconjugated ASO and cholesterol-conjugated ASO [30]. Part of the reason for the difference in activity between the cholesterol conjugates and the cholesterol unconjugates is the preferential accumulation of cholesterol conjugates in NPCs over hepatocytes in the liver. Watanabe et al. demonstrated that accumulation of cholesterol-conjugated ASOs was much greater in NPCs than in hepatic parenchymal cells as compared with unconjugated ASOs or GalNAc-conjugated ASOs [27]. Possibly providing support for the above explanation is a recent study by Donner et al., who showed that ASOs are inherently less active in NPCs than in hepatocytes [39].
Surprisingly, the effects of binding cholesterol to the 3′- and the 5′-terminus were comparable, differing from the finding of previous studies [26]. This provided a clue for how to optimize activity by introducing biolabile linkers. In addition, we noted a tendency for hepatic accumulation of
We utilized the ELOSA to quantify the amount of ASO in each tissue. In this assay, terminal modifications of ASOs could hamper the quantification result, because the ligands greatly affect the chemical properties of ASOs as well as enzymatic reactivity in ELOSA. However, it has been shown that ligands, such as GalNAc or cholesterol, connected to ASOs through phosphodiester linkers were immediately cleaved off, and the parent ASO was released within 48 h after dosing [27,40]. We determined the hepatic accumulation of
Renal accumulation of cholesterol–GalNAc dual conjugated ASOs
Next, to investigate renal accumulation, we quantitated levels of

Comparison of efficacy and renal accumulation of
Wolfrum et al. demonstrated that cholesterol-conjugated siRNAs preincubated with lipoproteins were delivered to liver cells through lipoprotein receptors, such as low-density lipoprotein receptor (LDLR) or Scavenger receptor class B member 1 (SR-B1) [20]. Nishina et al. reported that α-tocopherol-conjugated ASO admixed with lipoproteins had lower efficacy in the livers of LDLR-knockout (LDLR−/−) mice than those of wild-type mice [41]. In the present experiments, compared with wild-type mice, there was no difference in hepatic accumulation of cholesterol-conjugated ASO intravenously injected into LDLR−/− mice or apolipoprotein E-knockout (apoE−/−) mice without preincubation with lipoproteins (Supplementary Fig. S3). Thus, in our study, it is more likely that
In this study, we showed that our cholesterol–GalNAc dual conjugation strategy was clearly successful in reducing the dosage and altering the biodistribution of the parent unmodified ASOs, as well as reducing renal accumulation with no significant effect on hepatic gene-silencing efficacy. We plan to further test this dual conjugation strategy in preclinical models to evaluate the effects on the kidney in more detail.
Footnotes
Acknowledgments
This work was supported by Project Promoting Clinical Trials for Development of New Drugs (AMED) and the Platform Project for Supporting Drug Discovery and Life Science Research of the Japan Agency for Medical Research and Development (AMED).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.