
Abstract
Oligonucleotide therapeutics have emerged as a third distinct platform for drug discovery within the pharmaceutical industry. Five oligonucleotide-based drugs have been approved by the US FDA and over 100 oligonucleotides drugs are currently at different stages of human trials. Several of these oligonucleotide drugs are modified using the phosphorothioate (PS) backbone modification where one of the nonbridging oxygen atoms of the phosphodiester linkage is replaced with sulfur. In this review, we summarize our knowledge on receptor-mediated uptake of PS antisense oligonucleotides (ASOs) within different cell types of the liver—a privileged organ for the discovery of oligonucleotide-based therapeutics.
Introduction
O
A vast majority of the oligonucleotide drugs currently in preclinical and clinical development target genes expressed in the liver [7]. The liver plays a central role in metabolism, detoxification, and in the synthesis and secretion of important serum proteins, coagulation factors, hormones, and apolipoproteins. As a result, the liver is rich in gene targets for therapeutic intervention with oligonucleotide drugs, especially for diseases with strong genetic underpinnings. The liver comprises at least four distinct cell types. The hepatocytes represent the parenchymal cells and comprise almost 80% of the liver mass. The nonparenchymal cells (NPCs) include the liver sinusoidal endothelial cells (LSECs), the resident macrophage Kupffer cells, and hepatic stellate cells, which together comprise roughly 10% of the organ mass. Despite this, the NPCs represent 26% of total membrane surface, 58% of total endocytic vesicles, and 43% of the total lysosomal volume in the liver [8]. The different cells types within the liver express their own repertoire of cell surface proteins consistent with their roles in liver biology. The porous nature of the liver sinusoids provides access for macromolecular therapeutics, such as ASOs, to cell-surface acceptors on the different cell types within the organ.
Early work on the distribution of PS ASOs in animals showed that the liver and kidney accumulate higher levels of ASOs compared to other organs [9]. This work also established that PS ASOs do not accumulate uniformly across different cell types in the liver [9]. Instead, preferential uptake was observed in the NPCs, suggesting that these cells may possess unique molecular pathways for the uptake of PS ASOs [10]. In this review, we summarize recent advances in our understanding of the molecular pathways involved in the uptake of PS oligonucleotides within different cell types of the liver—an organ that occupies a privileged position for the discovery of oligonucleotide therapeutics.
Stabilin Receptors in LSECs
LSECs are a specialized class of endothelial cells that present unique structure and function in the liver compared to other endothelial cells in the body [11]. They contain numerous fenestrae or sieve plates that are bidirectional in fluid flow, allowing macromolecules in the plasma to directly contact hepatocytes [12]. LSECs represent about 15% of all cells of the liver and are responsible for roughly 45% of the pinocytic vesicles and 17% of lysosomal volume in the liver [13]. Consequently, they contain numerous vesicles involved with the internalization, transport, and degradation of material [14,15]. LSECs are the principle site for the systemic clearance of extracellular matrix [16] and internalize a host of natural ligands such as hyaluronan, chondroitin sulfates, immunoglobulins, advanced glycation end products, and collagen propeptides. They are also involved with the clearance of synthetic negatively charged molecules such as homogenous heparin [17] and, recently, PS ASOs [18]. The class H scavenger receptors, of which there are two family members (Stabilin-1 and Stabilin-2), have been found to be responsible for these activities.
Stabilin-1, also known as MS-1, FEEL-1, and CLEVER-1, is expressed in the sinusoidal endothelial cells of liver, spleen, lymph node, adrenal cortex, tonsil, and synovium of the joint [19,20] and in alternatively activated macrophages of the placenta [19], skin [21] and synovium [22]. In laboratory conditions, Stabilin-1 may be induced in human monocyte-derived macrophages by dexamethasone with or without IL-4 [23]. The residence time of Stabilin-1 on the cell surface is very low [24]; however, it binds cargo (acetylated low-density lipoprotein and PS ASOs) and shuttles it in the classical endocytic pathway that is Rab5 [25] and PI3K [26] dependent. A subset of Stabilin-1 receptors participates in trafficking to the trans-Golgi network as this receptor binds to Golgi-localized, gamma-ear-containing, adenosine 5′-diphosphate ribosylation factor-binding adaptors (GGA1 and GGA2) through an acidic domain within the intracellular cytoplasmic tail [23]. This acidic domain is not involved with endocytosis, but is essential for sorting interacting proteins such as SI-CLP [27]. Stabilin-1 also participates in transcytosis with the ligand, placental lactogen (PL), in which placental macrophages expressing high levels of Stabilin-1 maintain low levels of PL in the fetus compared to PL in maternal circulation [28]. The known ligands for Stabilin-1 are acetylated low-density lipoprotein [26], SPARC [29], PL [28], PS ASOs [18], phosphatidylserine [30], advanced glycation end products [31], and heparin [17].
Stabilin-2 [also known as the hyaluronic acid receptor for endocytosis (HARE), FEEL-2] is expressed in sinusoidal endothelium of liver, spleen, lymph node, bone marrow, and in specialized tissues of the eye, heart, brain, muscle, and kidney [32–35]. It is expressed as two isoforms (in human, 315 and 190 kDa) in native tissue [36] and stable cell lines expressing the recombinant protein [37]. The expression of Stabilin-2 is very high in native tissue, in which it is easily detected on the cell surface [37] with most of the receptor in intracellular compartments. Like Stabilin-1, Stabilin-2 undergoes classic clathrin-mediated endocytosis of ligands that may be inhibited by negative effectors for clathrin formation [25,37]. Both Stabilin-1 and Stabilin-2 have the same overall domain organization, which consists of 7 Faciclin-1 domains, 18–20 epidermal growth factor/EGF-like domains, a single transmembrane domain, and an X-Link domain, which is nonfunctional in Stabilin-1. The extracellular domain is 55% homologous with a 42% identity in contrast to the intracellular domains of both receptors, which are very different, in which Stabilin-2 does not contain a GGA adaptor, but does contain additional endocytic motifs that play a role in intracellular signaling that is ligand dependent [38]. The known ligands for Stabilin-2 include hyaluronan [39], several chondroitin sulfates [40], heparin [41], oxidized low-density lipoprotein [42], phosphatidylserine [43], and PS ASOs [44].
HEK293 cells overexpressing Stabilin-1 or either isoform of Stabilin-2 (190-HARE and 315-HARE) showed an enhanced uptake of an 125I-labeled PS ASO compared to the empty vector (EV) HEK293 cells (Fig. 1A). The uptake of the radiolabeled PS ASO in the Stabilin-expressing HEK293 cells could be competed using length- and chemistry-matched PS ASO, but only partially competed with a mixed-backbone ASO where some of the PS linkages in the parent ASO were replaced with POs (Fig. 1B). Interestingly, hyaluronic acid showed no competition, but heparin showed partial competition for uptake suggesting that heparin and PS ASOs may have partially overlapping binding sites on the receptor [18]. PS ASOs showed tight binding (KD ∼ 140 nM) to the ectodomain of 190-HARE using an enzyme-linked immunesorbent assay-like assay (Fig. 1C). It is likely that the large isoform of Stabilin-2 has similar binding affinity for PS ASOs based on previous experience with other ligands for the Stabilin receptors [37,45]. The ELISA-like method has not been developed for Stabilin-1 due to the lack of antibodies capable of capturing the Stabilin-1 ectodomain for plate-based assays. In the recombinant cell model, expression of Stabilin-2 correlates positively with enhanced knockdown of the targeted long noncoding RNA MALAT-1 (Fig. 1), suggesting that increased uptake allows for greater PS ASO escape from the endocytic pathway [18].
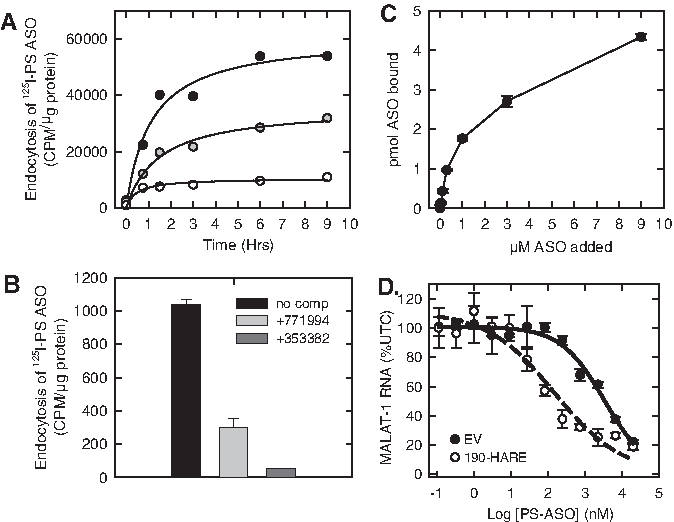
Stabilin interactions with PS ASOs.
PS ASOs bind to Stabilin receptors through a salt-bridge interaction to allow for internalization into early endosomal vesicles. While the majority of the internalized ASO is sorted to lysosomes (nonproductive pathway), a small percentage of the internalized ASO escapes the endolysosomal compartments and enters the cytosol and the nucleus, where it can bind its intended RNA target and exert antisense effects (productive pathway). PS ASOs showed an improved antisense activity in HEK293 cells expressing either Stabilin-1 or each isoform of Stabilin-2 compared to the EV control cells that did not express the receptor [18]. PS ASOs also showed higher accumulation in spleen and liver sinusoids of wt mice, but lower overall activity in the liver, compared to Stabilin-2 knockout mice. More recently, Rab5c (but not Rab5a or 5b) was shown to mediate the activity of PS ASOs after internalization in the Stabilin-2 HEK293 cells [46]. Rab5c interacts with early endosome antigen 1 (EEA1) to facilitate PS ASO transport from the early to the late endosome. After transport to the late endosome, Rab7a works in conjunction with lysobisphosphatidic acid (LBPA) to facilitate endosomal release of a small percentage of PS ASOs for bioactivity. Cell lines that are more amenable for activity with PS ASOs have a higher enrichment of LBPA, suggesting that this lipid along with the stabilizing protein Alix facilitates endosomal escape of PS ASOs [47].
Stabilin-mediated uptake of PS ASOs is an emerging area of research. Given that the Stabilin receptors are expressed in a wide array of organs and specialized tissues, it will be interesting to determine if these receptors are also involved in the productive uptake of PS ASOs in other cell types or tissues. There are clearly many unanswered questions at the interface of tissue biology and PS ASO endocytosis and bioactivity with regard to the Stabilin class of receptors
Asialoglycoprotein Receptor in Hepatocytes
The hepatocytes represent the major cell type of the liver and account for almost 80% of the organ mass. Hepatocytes synthesize and secrete serum proteins, coagulation factors, apolipoproteins, and hormones and are also involved in metabolism and detoxification [7]. Given the range of biological functions and plethora of gene targets expressed in hepatocytes, they represent a privileged cell type for the development of oligonucleotide therapeutics. The hepatocytes have direct access to blood flow through the sinusoidal fenestrae and express a range of cell surface receptors that localize in coated pits on the basolateral surface and are available for targeted delivery of therapeutic agents to this cell type.
The asialoglycoprotein receptor (ASGR) is a C-type lectin that is abundantly (∼500,000 copies/cell) [48] and almost exclusively expressed on hepatocytes in the liver [49–51] and regulates levels of plasma glycoproteins terminating with sialic acid α2,6 galactose and N-acetyl galactosamine (GalNAc) sugars [52,53]. There exists two homologous subunits of the ASGR (ASGR1 and ASGR2), which form a hetero-oligomeric complex with varying ratios (2–5:1) [54,55]. The ASGR clusters in coated pits of the basolateral membrane of hepatocytes where it is internalized by clathrin-mediated endocytosis [56,57]. Upon internalization, the ligand–receptor complex is transported to early endosomal compartments. The acidification that accompanies endosome maturation promotes dissociation of the ligand–receptor complex, after which the soluble ligand cargo is transported to lysosomes for degradation and the membrane-bound receptor is recycled to the plasma membrane [58]. Mice lacking the ASGR2 subunit are viable and fertile, but express the ASGR1 subunit at reduced levels [59]. Cells expressing ASGR1 alone are capable of binding and internalizing ligand [60], but substantial binding is dependent on the level of expression of the protein [61]. In contrast, mice lacking the ASGR1 subunit do not bind ligand and do not express ASGR2 on the plasma membrane [62]. The ASGR1 possesses a carbohydrate recognition domain for calcium-mediated sugar binding [63] and the cytoplasmic signal for binding clathrin adaptor proteins within coated pits [64,65]. Elegant work by Lee and colleagues showed that synthetic glycosides with branched tethers bind the ASGR with high affinity [66,67]. Binding affinity was dependent on the nature of the sugar (GalNAc > galactose), number of sugars (4 = 3 > 2 > 1), and the geometrical spacing between the sugar moieties [68]. The X-ray crystal structure of the ASGR1 ectodomain shows that the carbohydrate binding pocket is shallow and solvent exposed [63]. As a result, monovalent sugar ligands do not display high binding affinity for the ASGR and multiple interactions with the oligomeric receptor are required to enhance avidity [69].
Extensive structure activity relationships of synthetic multivalent GalNAc-ASO [70,71] and GalNAc-siRNA [72–74] conjugates have been reported. In these studies, GalNAc clusters with different linker lengths and configurations attached to the 5′-end of the ASO [75] were evaluated and shown to be essentially equivalent for enhancing ASO potency. Further studies revealed that two GalNAc sugars were sufficient and even a single GalNAc sugar can enhance ASO potency in cultured cells and in mice [76]. Receptor-binding experiments revealed that the PS backbone, charge, and single stranded character of the oligonucleotide can enhance the binding of mono-GalNAc designs for the ASGR [77]. The precise designs of multivalent GalNAc arrangement were less important compared to valency, and an oligonucleotide duplex with a single GalNAc sugar attached to each terminus of both strands exhibited the highest affinity. The receptor-binding observations could be rationalized by models where the relatively long oligonucleotide spacer helps the GalNAc sugars bind multiple receptors on the cell surface [77].
To determine if the interaction of PS ASOs with the ASGR in binding assays had functional consequences, both GalNAc-conjugated and GalNAc-unconjugated ASOs were evaluated in ASGR1 KO mice [77]. Significant reduction in antisense activity was observed for both the conjugated and unconjugated ASOs, supporting the hypothesis that the ASGR serves as a functional uptake pathway for PS ASOs in the liver. These observations were further strengthened by experiments with inducible ASGR1 HEK cells, where PS ASOs showed enhanced bulk uptake (Fig. 2A) and improved antisense activity (Fig. 2B) upon ASGR1 induction [78]. Interestingly, we observed reduced efficiency of internalization of GalNAc ASOs in uninduced HEK cells, which parallel our observations with other ligand-conjugated ASOs in HEK cells, which do not express the targeted receptor. A potential explanation is that nonspecific interactions of the ligand with cell surface proteins reduce the efficiency of the default “PS-uptake” pathways. Knocking down ASGR1 in wt mice resulted in a three-fold reduction in the potency of an unconjugated ASO targeting a hepatocyte-specific gene (Fig. 2C) [78]. Interestingly, the ASGR2 subunit was not involved in ASO uptake in cells or in mice.
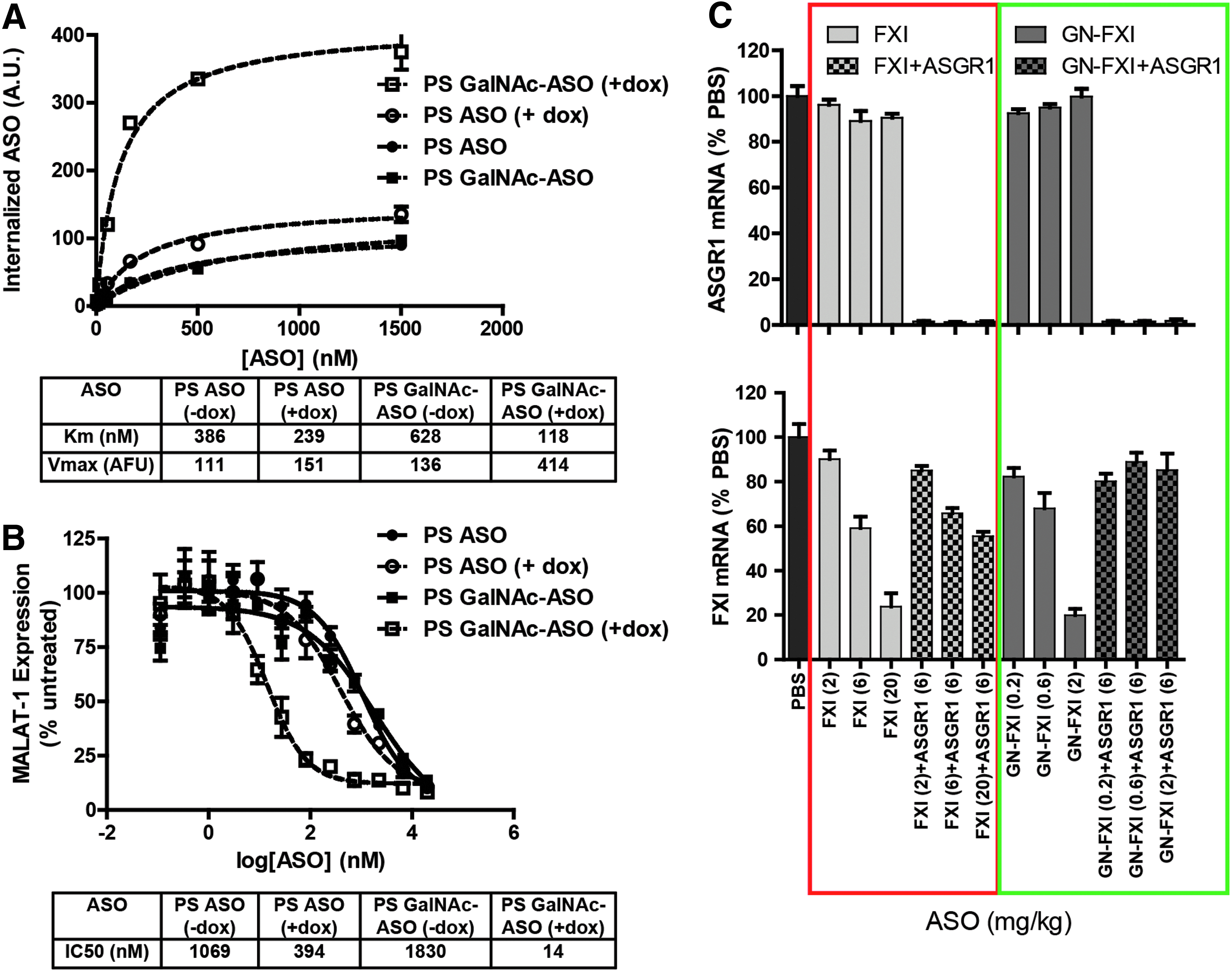
ASGR1 contributes to uptake and activity of parent PS ASOs both in vitro and in vivo.
Scavenger Receptor A in Kupffer Cells
Kupffer cells are the resident macrophage cells of the liver and together with the LSECs, account for the bulk of ASO uptake in the liver [79,80]. Early work showed that uptake of PS ASOs in the liver could be competed with polyanionic molecules such as polyI and polyG, which are known ligands for receptors such as scavenger receptor A (SRA) [10]. Subsequent work in SRA knockout mice showed no difference in bulk accumulation or antisense activity of PS ASOs in the liver, except a small, but significant reduction (∼25%) in the accumulation in Kupffer cells [81]. Further work in peritoneal macrophages suggested the existence of high-affinity/low-capacity and low-affinity/high-capacity mechanisms for ASO uptake in macrophage-type cells. A 50% decrease in high-affinity uptake with no change in the low-affinity uptake between macrophages isolated from SRA knockout and wt mice was observed. This study concluded that SRA could be one of several molecular pathways responsible for the uptake of PS ASOs in Kupffer cells. A recent study also suggested that some classes of chemically modified oligonucleotides form nanoparticles, which are taken up into macrophages by SRA [82]. However, no direct interaction with the receptor was convincingly demonstrated. Given the spectrum of scavenger-type receptors expressed on macrophages, it is likely that PS ASOs are taken up in these (and other) cell types by direct interactions with these receptors and as ASO complexes with serum proteins like α-2-macroglobin. This hypothesis is supported by recent work where a two-fold enhancement in ASO potency in the liver was observed in α-2-macroglobin knockout mice [83].
Using Engineered Cells to Understand Receptor-Mediated Uptake of ASOs
Identifying appropriate in vitro cellular systems to identify receptors involved in uptake of single-stranded PS ASOs has proved challenging. From our collective experience over several years, most mammalian cells in culture will internalize PS ASOs without the aid of transfection or delivery agents. PS ASOs adhere to cell surface proteins by virtue of their protein binding properties. One study estimated 200,000 binding sites for PS ASOs on the surface of K562 cells [6]. Following binding to the cell surface, PS ASOs are internalized and delivered into endolysosomal compartments by specific endocytic processes or during the course of general membrane turnover. Only a subset of cell types and immortalized cell lines, however, shows good functional uptake where ASO internalization is accompanied by reduction of the cellular RNA target of the ASO. Furthermore, there appears to be little correlation between bulk uptake and functional uptake as illustrated by our studies with HepG2 cells to study uptake of GalNAc-conjugated and GalNAc-unconjugated PS ASOs [78].
Given that PS ASOs can bind to a large number of proteins on the cell surface and in the extracellular matrix [84], manipulating levels of a single cell surface protein to determine its role in PS-mediated uptake can often at times lead to inconclusive results. Extrapolating results from cultured primary cells to in vivo models can also be misleading as primary cells often change/lose expression of surface receptors in culture. Similarly, the cell surface proteome of a given cancer cell line is also likely to be different from other cancer or primary cells. Given these issues, we utilized an HEK293 system to probe the role of individual receptors in uptake of PS ASOs, as they represent a well-defined, easily manipulated in vitro cell model. HEK293 cells appear to have limited basal capacity for uptake of PS ASOs, providing a low background for the detection of increased uptake by heterologously expressed membrane proteins. Heterologous overexpression allows one to examine the role of a given receptor or to compare different receptors for their ability to internalize ASOs in a productive manner against an isogenic background. For example, we have compared the relationship of uptake versus activity for both PS-mediated and ASGR1-mediated ASO uptake in ASGR1-expressing HEK cells (Fig. 3). We found similar levels of uptake at the respective IC50 of parent and GalNAc-conjugated ASOs, suggesting ASGR1-mediated uptake is not intrinsically more productive than PS-mediated uptake.

Uptake of parent and GalNAc-conjugated ASOs at their respective IC50s is similar. Interpolated values from flow cytometry uptake experiments with ASGR1 HEK cells were used to compare relative uptake of parent and GalNAc-conjugated ASOs at their respective IC50s.
Conclusion
The liver represents an important organ for the development of oligonucleotide therapeutics. PS ASOs are efficiently taken up by the liver after systemic injection (Fig. 4). However, the majority of the ASO accumulates within the NPCs [79], while the gene targets of therapeutic interest are often expressed in hepatocytes [7]. As a result, higher doses of oligonucleotides are required to saturate ASO-uptake pathways in the NPCs to achieve effective gene silencing in hepatocytes [80]. Early work had implicated scavenger receptors as being responsible for uptake of PS ASOs within NPCs of the liver. However, the precise identity of the receptors responsible for uptake of PS ASOs within the different cell types of the liver remained largely unknown. Recent work from our laboratories established that the Stabilin receptors are responsible for uptake of PS ASOs into the LSECs. Shifting ASO distribution from NPCs to hepatocytes by ASGR-mediated delivery enhances potency 10- to 30-fold in mice and man [85]. Interestingly, our work also suggests that the ASGR is responsible for the uptake of a significant fraction of PS ASOs into hepatocytes. A broader understanding of molecular pathways for the uptake of PS ASOs in other cell types can help design better ASO drugs for treating diseases in tissues beyond the liver.
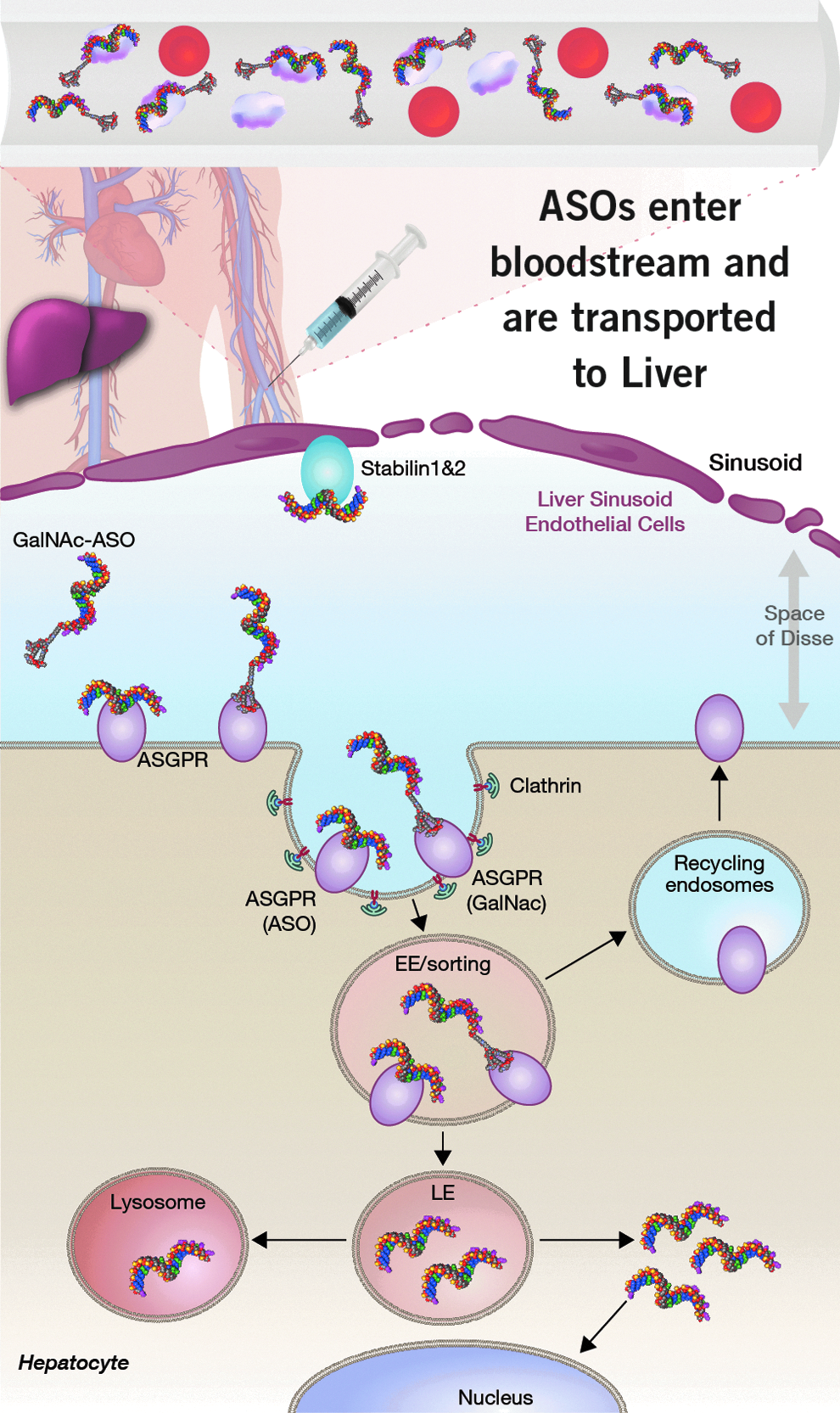
The PS backbone promotes binding to plasma proteins and facilitates ASO distribution to the liver and other tissues from the site of injection. ASOs are efficiently internalized into LSECs by the stabilin receptors by interactions with the PS backbone. In contrast, ASOs are internalized less efficiently into hepatocytes by interactions with the PS backbone. Targeted delivery of ASOs into hepatocytes by the ASGR using GalNAc-ASO conjugates improves potency for inhibition of gene targets expressed in hepatocytes. LSECs, liver sinusoidal endothelial cells.
Footnotes
Acknowledgments
The authors thank Tracy Riegle for graphical assistance and Drs. Frank Bennett and Hans Gaus for useful discussions.
Author Disclosure Statement
The authors declare no financial conflicts of interest. M.T., A.J.D., T.P.P., E.E.S., and P.P.S. are employees of Ionis Pharmaceuticals, Inc.