
Abstract
Increased interest and insights gained by researchers on the roles of endothelial cells in the pathophysiology of cancer, inflammatory, and cardiovascular diseases have led to the design of pharmacological interventions aimed at the endothelium lining in the diseased sites. Toward this end, we used established brain microvascular endothelial cell lines mouse (bEND3), human (hCMEC/D3), and Toggle Cell-SELEX to identify a species cross-reactive, endothelial cell-internalizing aptamer R11-3. This 2’F-modified RNA aptamer is specific for endothelial cells as no internalization was seen with cells of nonendothelial origin. R11-3 was truncated in size, and its potential in endothelial targeted therapeutics was established using VEGFR2 targeting long interfering RNA (liRNA) aptamer chimera. Due to its specificity for both mouse and human endothelial cells, we believe that this aptamer not only fits for development of endothelial targeted drug development for human diseases but is also suitable for preclinical evaluation in mice.
Introduction
E
Endothelial cell targeting carrier molecules that recognize endothelial cell surface proteins and subsequently get internalized are promising candidates for the development of therapeutics and imaging systems in disease states. Toward this end, in the past few years, some efforts have been focused on the development of endothelial-targeted aptamers [3–5]. Aptamers are short single-strand (ss) DNA or RNA oligonucleotides that fold into three-dimensional secondary structures and bind to a target with high affinity and specificity. Their nanometer size range and feasibility for chemical conjugation make them very efficient drug carrier molecules [6]. Cell-internalizing aptamers are well suited for targeted delivery of drugs, miRNA, and siRNAs. Other than known cell surface receptors, the aptamers can also be targeted for whole cells by Cell-SELEX tailored for the enrichment of cell-internalizing aptamers [7]. While some of them act as general cell carriers [8–10], others targeting disease-associated receptors are specific to cells. The most studied aptamer for endothelial use is the human transferrin receptor (TfR) aptamer [11]. This aptamer has been shown to internalize endothelial cells and deliver therapeutic miRNA [10]. Although, human and mouse TfR have 80% homology, this aptamer failed to recognize mouse endothelial cells. Similar aptamer prepared earlier for mouse TfR failed to react with a human ortholog [12], suggesting that, even with highly homologous target proteins, species cross-reactivity of the aptamer cannot be guaranteed, unless the selection procedure is designed for it.
In another report, a blood–brain barrier (BBB) permeable aptamer was identified by in vivo Cell-SELEX, which also internalized mouse cerebral microvascular cells [13]. Whether this aptamer can recognize human cerebral endothelial cells or can pass through human BBB is something which needs to be investigated. We believe that species cross-reactivity is a huge limiting factor in the preclinical evaluation of therapeutic ligands, especially when they are directed to cells like these, for which xenografts are not possible. This is more important for aptamers against novel therapeutic targets, whose clinical association is not well established or where the target proteins are not known. While it is an important concern, such ligands that demonstrate cross-reactivity to rodent or nonhuman primate orthologs are rare.
To address this caveat, in our current study, we developed a mouse/human cross-reactive, nuclease-resistant aptamer for endothelial cells. The aptamer was selected by cell-internalizing toggle Cell-SELEX [14] that involved a stringent selection of human and mouse cerebral endothelial cell-internalizing sequences. Using this approach, we identified an aptamer R11-3 that could specifically internalize both mouse and human endothelial cells. The aptamer was minimized to 39nt in size and its potential in the endothelial targeted delivery of vascular endothelial growth factor receptor (VEGFR2) targeting long-interfering RNA (liRNA) was demonstrated using an aptamer-liRNA chimera.
Materials and Methods
Cell culture
Human cerebral microvascular endothelial cell line hCMEC/D3 was obtained from P-O Couraud, INSERM, France [15]. Human vascular endothelial cells (HUVEC) were obtained from the American Tissue Culture Collection (ATCC). hCMEC/D3 and HUVEC were cultured in EGM-2 Bullet Kit (CC-3156 & CC-4176) (Lonza Biologics, Walkersville, MD). Human brain microvascular endothelial cells (HBMEC), Catalog No: SC1000 were purchased from Cell Systems (Kirkland, WA) and cultured in CSC Complete Medium (4Z0-500) in accordance with the supplier's recommendations. Bend3 mouse endothelial cell line was obtained from ATCC and grown in DMEM supplemented with 10% fetal calf serum (Invitrogen, CA). Cells were cultured in humidified conditions at 37°C in a 5% CO2 atmosphere.
Aptamers and siRNAs
Previously reported BBB-penetrating A15 aptamer (GGGAGGACGAUGCGGCGUA UUGCGCGAGGAUUAUCCGCUCAUCGUUGUUGUUGUGCAGACGACUCGCCCGA) [13], TfR aptamer C2.min (GGGGGAUCAAUCCAAGGGACCCGGAAACGCU CCCUUACACCCC) [11] were prepared by in vitro transcription. Aptamer controls LSQ2 and LSQ5 are random oligonucleotide controls obtained from the N40 library by TA cloning. Coding DNA strands containing an upstream T7 promoter were purchased from Integrated DNA technologies (IDT, Coralville, IA) and transcribed using DuraScribe T7 Transcription Kit (Epicentre Biotechnologies, Madison, WI). RNA was purified on urea-PAGE, and quality and size were confirmed on the gel. VEGFR2-targeting siRNAs (sense: GUCCCUCAGUGAUGUAGAAdTdT) and liRNA (sense: GUCCCUCAGUGAUGUAGA A
Endothelial cell-internalizing toggle cell-SELEX
The starting 2′-F modified RNA library was prepared by in vitro transcription of an N40 DNA using a DuraScribe® T7 Transcription Kit. A total of 2.5 nM of this RNA library was used for Cell-SELEX. In brief, the RNA library was denatured in binding buffer (Dulbecco's PBS buffer containing 4.5 g/L glucose and 5 mM MgCl2) for 5 min at 95°C and gradually cooled down to RT for 10 min. Each hCMEC/D3 and bend3 cell line was grown as a monolayer and was preblocked with 100 μg/mL of ytRNA in serum-free culture medium at 37°C for 15 min. Following removal of the blocking medium, the cells were further incubated with 2.5 nM of the 2′F RNA library with 100 μg/mL of ytRNA as competitors for 60 min at 37°C. The unbound sequences were removed by two brief washes with PBS. Cell-surface bound sequences were removed by 10 min treatment with Trypsin-EDTA. The cell pellet was washed twice with chilled PBS and lysed using a TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA was extracted and reverse-transcribed using 50 pM of N40 reverse primer (5′-AGATTGCACTTACTATCT-3′) with ImProm-II™ Reverse Transcription System (Promega, Madison, WI), and PCR amplified using N40 reverse and forward primer [containing T7 promoter (underlined) (5′-
For a stringent selection of the cell-internalized RNA, in later cycles, trypsin-EDTA treatment was followed by high salt-acid stripping as described before [16]. In brief, the cell pellet was washed with 500 μL of a chilled solution of 0.5 M NaCl and 0.2 M acetic acid for 2 min on ice. Cells were washed once with 1 mL of chilled PBS and then lysed using TRIzol reagent. SELEX was continued for a total of 13 rounds. Details of each round are provided in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/nat). SELEX enrichment was quantified by real-time qPCR. In the 11th and 13th round, sequence pool was cloned using a TA Cloning Vector Kit (RBC Bioscience Corp. Taiwan). The 25 clones from each round were sequenced and analyzed for consensus using MultiAlign software [17]. RNAstructure V5.8.1 was used to predict the secondary structures of aptamers. Truncated versions of R11-3 DNA oligo coding strands (IDT) containing an upstream T7 promoter binding sites were transcribed in vitro to obtain 2′-F modified truncated versions of the aptamer. The binding affinity of R11 and truncated R39 aptamer was seen at a concentration of 1–100 nM at 37°C. Saturation curves and Kd values were obtained using Sigma plot v10.0.
Internalization assay
Cells grown in 12-well plates were incubated with 50–100 nM of folded aptamer sequences in the respective serum-free growth media for 30–60 min at 37°C as indicated. In case of metabolic inhibition, cells were pretreated with 50 mM of 2-deoxyglucose and 10 mM of NaN3 in PBS for 30 min and further incubated with the aptamers in the same buffer. Unbound aptamers were washed with PBS, and the cell-bound RNA was removed by treatment with 0.25% trypsin-EDTA followed by high salt–acid stripping. Cells were lysed using isol-RNA lysis reagent (5 PRIME, Inc., Gaithersburg, MD) 500 ng of total RNA and the respective input aptamer standards were then reverse-transcribed with 50 pM of reverse primer using the ImProm-II™ Reverse Transcription System (Promega), according to the manufacturer's protocol. Aliquots of the cDNA reaction mixture were analyzed by qRT-PCR using input aptamer cDNAs as standards and specific primer pairs.
Microscopic analysis
For microscopic analysis, R11-3 aptamer was labeled with TAMRA using a reverse primer with the fluorescent probe at 3′ end. In brief, TAMRA labeled reverse primer was mixed with gel purified in vitro transcribed R11-3 RNA, at a molar ratio of 1:1, in the RNA duplexing buffer (60 mM KCl, 6 mM HEPES-pH 7.5, 0.2 mM MgCl2). The annealing reaction was performed on a thermocycler with the settings: 95°C initial denaturation, gradient step down of 2.5°C/min to 37°C, hold for 30 min at 37°C, and finally cooled to 25°C. The annealed products were run on 10% native PAGE and annealing of the fluorescent tagged primer to R11-3 RNA was confirmed by visualization on a UV illuminator without Ethidium Bromide staining. Cells grown on glass bottom Petri-dishes were incubated with 100 nM of 3
Aptamer-RNAi chimera preparation and transfection
R39-siRNAsense (
Quantitative real-time PCR
mRNA was extracted using an Isol-RNA Lysis Reagent (5 PRIME, Inc.). 500 ng of RNA was used as a template for cDNA synthesis, using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), according to the manufacturer's protocol. Aliquots of the cDNA reaction mixture were analyzed for VEGFR2 (forward: GGAAGCTCCTGAAGATCTGT, reverse: GAGGATATTTCGTGCCGC) and β tubulin (forward: GACCAACCGTACCATCCAGT and reverse: CACGTTTGGCATACATCAGG) by quantitative real-time PCR on a Step-One real-time PCR machine (Applied Biosystems). VEGFR2 mRNA was normalized with β-tubulin, and quantities relative to controls are shown.
Statistical analysis
All experiments were performed at least in triplicates for reproducibility, and the data are represented as mean +SE of three or more independent experiments. Student's t test with a two-sided alpha of 0.05 was used for comparison of the differences between treatment groups.
Results
Species cross-reactive endothelial cell-internalizing aptamer selection
To select a species cross-reactive endothelial cell-internalizing aptamer, Cell-SELEX was performed with established human and mouse microvascular endothelial cell line, hCMEC/D3, and bEND3 (Fig. 1A). To ensure that the SELEX favors selection of ligands for both the target cells, the first round comprised of selection against both cell types separately, following which the output RNA from both the SELEX were pooled in equimolar quantities and used for next round. After that, Toggle SELEX was performed with hCMEC/D3 or bEND3 in alternate rounds. To remove the cell membrane-bound sequences from the cell-internalized fraction, trypsin EDTA and high salt–acid stripping were preferred over standard RNAse treatment protocol [8], as the latter was found to be harsh for endothelial cells, often leading to the degradation of the internalized fraction.
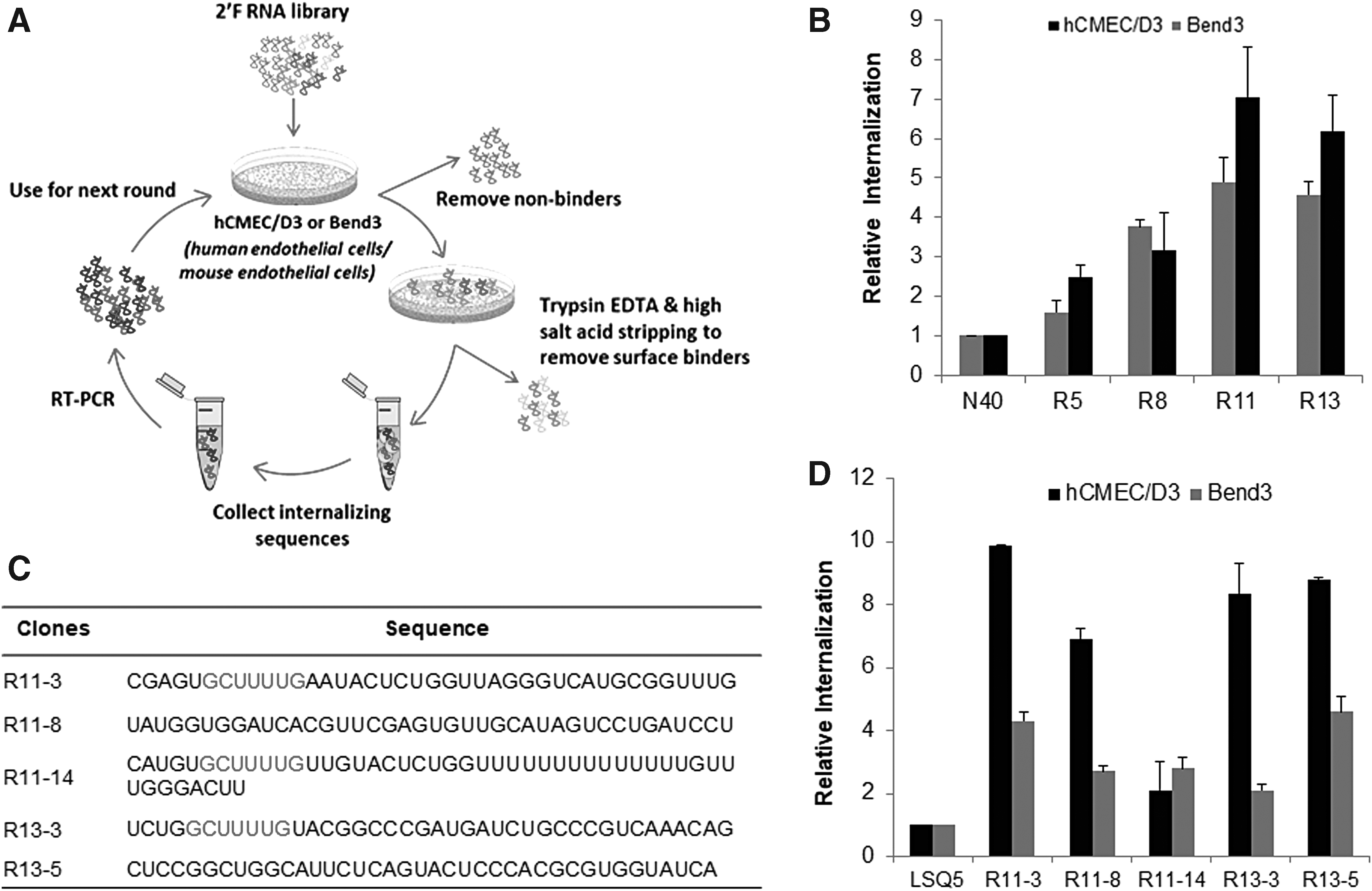
Toggle cell-internalizing SELEX for endothelial cell-internalizing aptamers.
The enrichment of binders during the selection process was measured using qRT-PCR. The starting N40 library and the 5th, 8th, 11th, and 13th round pools were tested for binding to hCMEC/D3 and bEND3 cells. Both the cells showed increased internalization of RNA with increasing cycles when compared to the starting library (Fig. 1B). As the 11th and 13th round pool showed significant enrichment and saturation in internalization, the SELEX process was discontinued, and the RNA pools from these two rounds were cloned and sequenced. Sequences were aligned into families according to sequence homology. As shown in Fig. 1C, five distinct consensus sequences were identified, and a seven nucleotide long common motif (GCUUUUG) was seen in three of these sequences; R11-3, R11-14, and R13-3 (marked in red), indicating toward a common target interaction domain. Internalization studies were performed with these sequences using real-time qPCR and their uptake by hCMEC/D3, and bEND3 was found to be 2–10 times higher than that of the initial library (Fig. 1D). Aptamer internalization was also confirmed by fluorescence microscopy using a TAMRA-labeled reverse primer annealed to the aptamer sequences (Supplementary Fig. S1A, B). Among the five selected sequences, aptamer R11-3 showed the maximum internalization in both qPCR and imaging and hence was chosen for further characterizations.
R11-3 internalizes exclusively in endothelial cells and outperforms some known endothelial internalizing aptamers
R11-3 aptamer was selected against immortalized endothelial cells. Therefore, it was important to confirm that the aptamer also recognized primary endothelial cells. To validate this, we checked the internalization of the aptamer in HBMEC and HUVEC cells using fluorescence microscopy. The aptamer was labeled with TAMRA using reverse primer and incubated with cells at a concentration of 100 nM for 30 min. DIC and TAMRA overlay images show uptake of R11-3-TAMRA by all the four endothelial cell types tested in this study (Fig. 2A). Most of the fluorescence was localized in the perinuclear regions of the cells. No internalization was seen with the LSQ2 control sequence. Next, we tested whether the aptamer internalization is specific for endothelial cells. For this, we chose a panel of cell types with diverse cell origins; NIH3T3 (fibroblast), HPDE (pancreatic ductal), THP1 (monocytes), NHA (astrocytes), HEK293 (embryonic kidney), and HaCaT (keratinocyte). R11-3 internalization was not seen with any of these cells with microscopy (data not shown). We confirmed our observation with qRT-PCR, and Fig. 2B confirms that aptamer R11-3 internalizes specifically to endothelial cells.
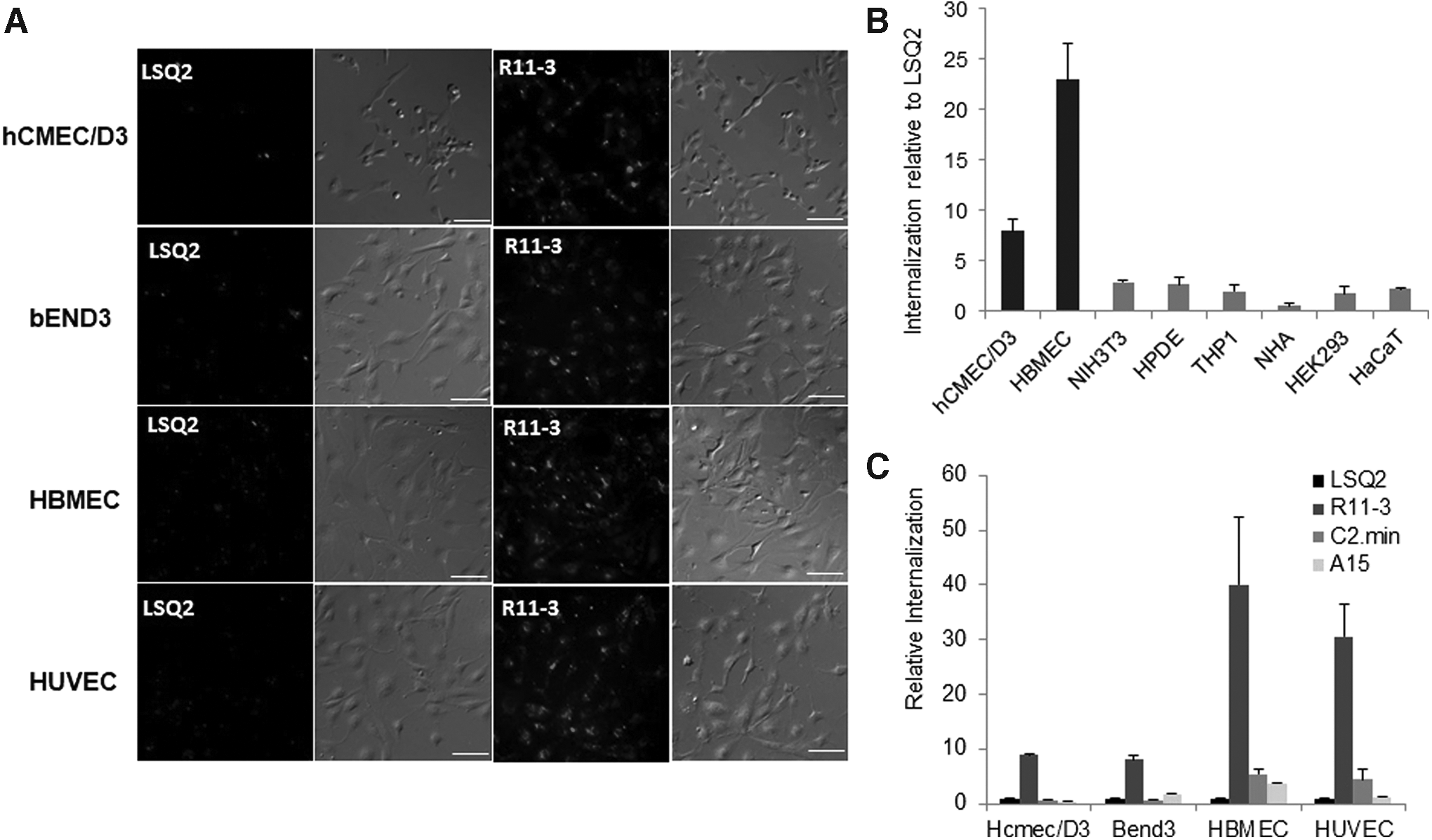
R11-3 internalization is specific for endothelial cells and is more efficient than other endothelial aptamers.
We also compared our R11-3 aptamer with two published 2’F modified RNA aptamers; the well-known human transferrin-binding aptamer C2.min [11] and A15 mouse BBB penetrating aptamer [13]. Aptamers were prepared by in vitro transcription and incubated with the mouse and human endothelial cells for 60 min. Internalized aptamers were quantitated by qRT-PCR using specific primer pairs. Figure 2C shows the amounts of internalized aptamer relative to LSQ2 control. Against our expectations, both transferrin and A15 aptamer showed very low internalization. Nevertheless, R11-3 showed significant uptake in all the four cell types.
Aptamer R11-3 structure-based truncation identified 39 nt miniaturized aptamer, R39 with a binding affinity similar to the full-length form
To further use the aptamer R11-3 in any targeted delivery application and easy chemical synthesis, we tried reducing its size with minimal loss in target binding. The predicted secondary structure of full-length R11-3 (78nt) shows folding into a two-way stem loop structure, one containing the upstream primer binding region and a part of the random sequence (nt 3-41) and the other containing the downstream primer binding region and the remaining of the random sequence (nt 44-78) (Fig. 3A). Using this structural information, we prepared two smaller variants, R11-3(3-41) and R11-3(44-78). Also, R11-3 random (19-59), and primer-deleted sequences R11-3-up truncated (19-78), and R11-3-down truncated versions (1-59) were prepared and tested for their internalization by hCMEC/D3 cells.

Aptamer size minimization and affinity measurement.
Figure 3B shows that the random aptamer region alone has no internalization activity. A significant loss in internalization was also seen with a variant that lacks the upstream primer binding sequence. Altogether, this shows that upstream primer region of the aptamer is important for its activity. Downstream primer binding region had no role in aptamer internalization. R11-3(3-41) retains the cell internalization activity, while R11-3(42-78) is completely dispensable. The enriched motif (GCUUUUG) is present in the loop 1 of the active variant R11-3(3-41), suggesting that this may be the target recognition domain. The upstream primer binding sequence makes the rest of the loop 1, signifying that the minimal binding aptamer structure, hereafter referred as R39, retains the constant region of the sequence. This generally occurs if very high stringencies within the selection process are applied, such that even the fixed primer binding sites may be directly included in the active functional sequences, a phenomenon that is likely to occur in Toggle-SELEX [19].
Next, we tested binding affinity of the R11-3 and the truncated aptamer R39 over a range of concentrations, Fig. 3C shows saturation at a concentration higher than 20 nM and a dissociation constant of 3.4 ± 1.2 nM and 1.2 ± 0.2 nM, respectively.
Aptamer R39 is internalized via endocytosis and efficiently delivers cargo inside the cell
Binding and internalization studies show that 21% ± 2.2% of the bound aptamer enters the cell in 30 min. Uptake kinetics of over the course of 60 min show maximum uptake within 30 min of incubation. Further incubation did not increase the amount of R39 in the cells (Fig. 4A). Also, no significant internalization was observed at 4°C (Fig. 4B). Temperature-dependent uptake, perinuclear localization, and saturation beyond a particular concentration altogether indicated an energy-dependent endocytic uptake. We confirmed this using metabolic inhibitors that result in cellular ATP depletion. Cells arrested in this state showed significantly reduced uptake of R39 (Fig. 4B), confirming aptamer internalization by endocytosis.

R39 internalizes by endocytosis and can be used for targeted delivery.
Next, we tested the cargo carrying capacity of the aptamer inside the endothelial cells. As a proof of concept, we tried to deliver streptavidin-coated magnetic beads using biotinylated R11-3 aptamer. Aptamer-bead complex incubated with hCMEC/D3 resulted in bead internalization into the cells, whereas no bead binding or internalization was seen in case of the LSQ2-bead complex (Fig. 4C).
R39-mediated delivery of VEGFR2 targeting liRNA reduces target gene expression
To further demonstrate the use of R39 aptamer in endothelial targeted therapeutics, we tried to deliver RNAi triggers targeting VEGFR2 mRNA. Biological and preclinical evidence suggests that the blockage of VEGFR2 could be a promising strategy to inhibit angiogenesis [20]. In addition to the standard aptamer-siRNA chimera that uses the conventional 19 + 2 siRNA, we also prepared aptamer-liRNA (Fig. 5A) that is based on the long interfering dsRNA design, earlier published by our group [21,22]. The 5′ end 19 nt of the AS strand was identical to the corresponding 19 bp siRNAs, and the 19 nt extensions complementary to the target mRNA were made at the 3′ end to ensure annealing with other siRNA units. The 38 nt sense strand was designed to have both the 19 nt 5′ end and the 19 nt 3′ end complementary to the corresponding AS strand. Thus, annealing of two strands resulted in the formation of multimeric long dsRNAs nicked at every 19 bp. liRNA and R39-liRNA chimera were annealed, and their quality and size were confirmed on PAGE (Fig. 5B).
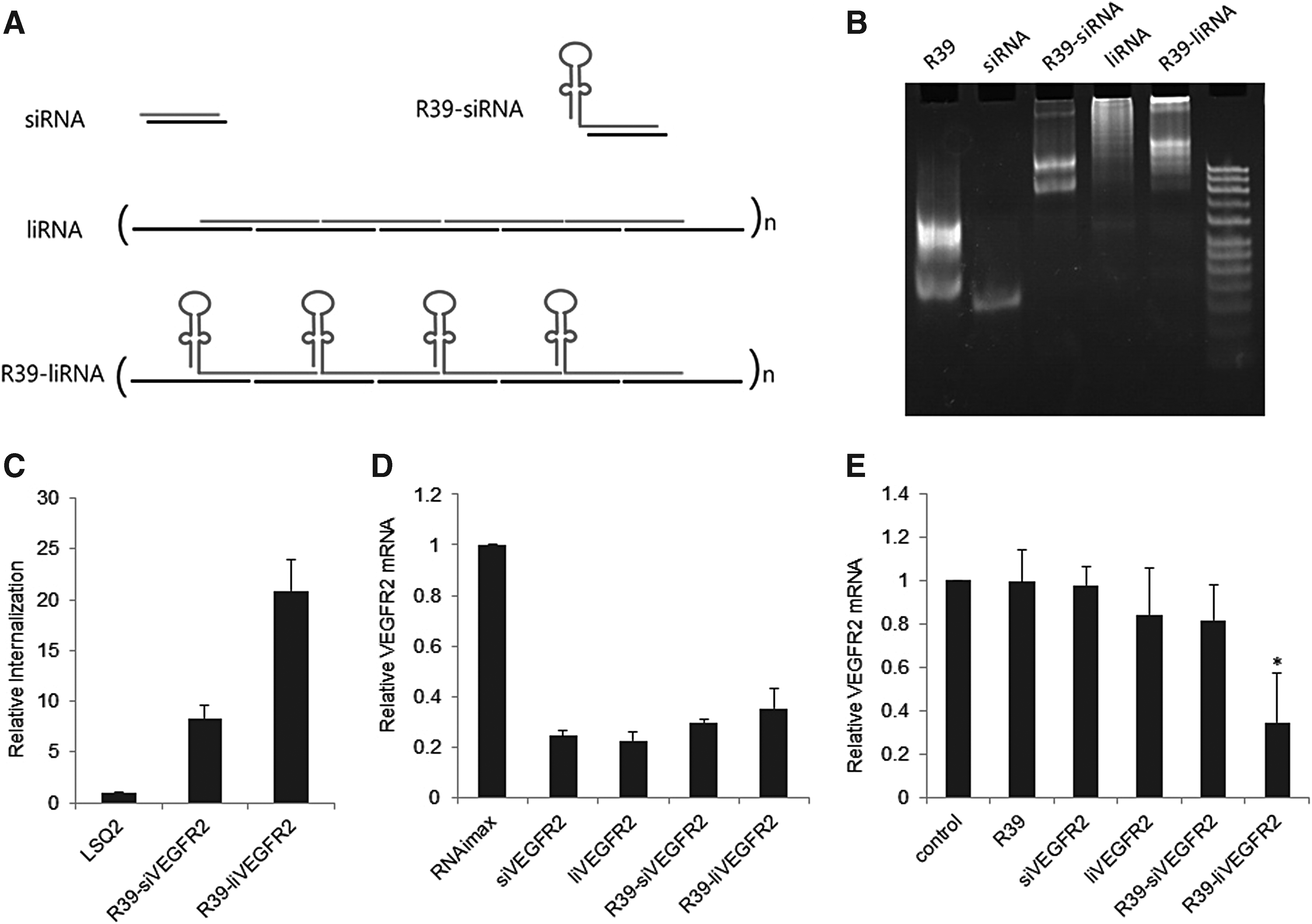
R39-VEGFR2 liRNA chimera preparation and its targeted delivery to endothelial cells.
The concatamer format of liRNA allows incorporation of multiple aptamer and siRNA units, alternating with each other in a long polymeric form. We believed that this arrangement would not only increase the valency of the aptamer for superior target binding but could also deliver multiple siRNA molecules in a single internalization event. Internalization assay with R39-siRNA and -liRNA confirmed our hypothesis. Aptamer-liRNA chimeras could internalize hCMEC/D3 cells with more than two-fold higher efficiency (Fig. 5C). We then tested the mRNA knockdown efficiency of the chimeras in comparison to the siRNA/liRNA alone. Cells transfected with siRNA and liRNA resulted in 75% reduction in VEGFR2 mRNA levels (Fig. 5D). Although not significant, some loss in silencing was observed with the respective chimeras, but overall, both the chimeras could efficiently reduce target mRNA levels when delivered with lipid-based transfection reagent. However, in the absence of transfection reagent, aptamer-siRNA chimera failed to achieve any significant target knockdown. In contrast, a 65% knockdown could still be observed with the liRNA chimera (Fig. 5E). The duration of treatment was limited to 6 h to avoid immune-stimulatory responses due to the polymeric nature of RNAs. This shows that such multimeric format of aptamer-siRNA chimera with multivalent nature and better aspect ratios provide superior delivery and mRNA knockdown efficiency.
Discussion
Cell-internalizing aptamers have tremendous potential for theranostics. They not only recognize the target cell but can also cross the cell membrane to reach the cytosolic compartment, where the payload is supposed to be released. Furthermore, with a well-defined selection strategy and proper partitioning, Cell-SELEX can be driven toward the identification of aptamers in accordance to the unique requirement. Altogether, this has revolutionized the field of targeted drug delivery [23,24]. Taking advantage of the flexibility of the Cell-SELEX procedure, in this study, we combined toggle Cell-SELEX and cell-internalizing SELEX and identified a human/mouse cross-reactive cell-internalizing aptamer for endothelial cells. Although selected against human and mouse cerebral microvascular cells, our R11-3 aptamer could also recognize HUVEC. No internalization was seen in cells of nonendothelial origin, suggesting that aptamer is specific for endothelial cells, and its use is not restricted to cerebral microvascular endothelial cells alone. Whether this aptamer can transcytose cerebral endothelial cells and penetrate blood–brain barrier is something which needs to be investigated.
R11-3 enters the cell via endocytosis and could efficiently deliver magnetic beads of more than 2 μm in size, suggesting that the aptamer may be suitable for functionalization of nanoparticles and other large molecules. For easy chemical synthesis and downstream applications, we removed the unwanted sequences from the aptamer and reduced it to 39 nt in length while maintaining the binding affinity and internalization efficiency. Finally, to demonstrate the use of R39 in endothelium targeted delivery, we chose VEGFR2 as a therapeutic target and designed siRNA against it. For therapeutic molecules such as siRNA and miRNA, where delivery is the biggest hurdle, aptamer-mediated delivery has shown promising results [25–27]. Especially, in case of RNA aptamers, due to similar nucleic acid chemistry designing aptamer-siRNA chimeras is straightforward. However, our initial attempts with R39-mediated delivery of siVEGFR2 failed to achieve any mRNA knockdown. For efficient target mRNA knockdown a significant copy number of siRNAs should be able to reach cytosol. In our past experiences, we have seen that RNAi efficiency of aptamer-siRNA chimeras is often suboptimal and inconsistent as they lack an endosomal release accomplice for cytosolic release [28,29]. To some extent, absence of a proper endosomal release can be compensated by increasing the amount of RNAi being delivered to the cell. To achieve this goal, we developed an aptamer RNAi chimera using liRNA, earlier reported by us. The concatamer design of liRNA allowed us to incorporate R39 alternating with siVEGFR2 in a polymeric form. With this format, we could not only make multivalent aptamer for superior delivery but could also deliver multiple copies of siRNA. A significant mRNA knockdown could be achieved with this polymeric chimera. In an earlier report, similar aptamer-siRNA polymers were constructed using Muc1 DNA aptamer and GFP targeting siRNAs [30]. In this experimental design, multimeric antisense was prepared via thiol conjugations, and Muc1 sense strand DNA extension was annealed onto it. However, the aptamer-mediated delivery of this siRNA polymer failed to achieve any target knockdown, which could be due to the DNA-RNA hybrid chemistry or poor endosomal escape.
As the aptamer R39 was selected by Cell-SELEX, its target identity is unknown. Identification of the target protein will further help in ascertaining the use of this aptamer in vascular diseases. Work in this regard is underway. We believe that our aptamer is more efficient than other aptamers reported for similar use. Also, its human and mouse cross-reactivity not only makes it suitable for preclinical screening but also for direct use in clinical studies of targeted vascular therapeutics.
Footnotes
Acknowledgments
This work was supported by Basic Science Research Programs through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF 2013R1A1A2062908, NRF 2017R1D1A1B03036001) and a Global Research Laboratory grant from the Ministry of Education, Science, and Technology of Korea (no. 2008-00582). We also acknowledge and thank Dr. Pierre-Olivier Couraud for providing us with the hCMEC/D3 cell line for this study.
Author Disclosure Statement
All authors have no competing financial interests to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.