
Abstract
Translation of in vitro transcribed messenger RNA (mRNA) is known to be compromised by cell's innate immune responses. Herein we show that when mRNA encoding nonstructural protein 1 (NS1), an immune evasion gene derived from influenza A virus, is co-delivered with mRNA encoding green fluorescent protein (GFP), higher GFP expression can be observed in four different interferon competent cell types within 6 h, indicating NS1's wide host range property and rapid counter response to the cells' innate immune response. Enhanced mRNA translation correlates with reduced interferon production in all tested cell types and substituting a small portion of luciferase mRNA with NS1 mRNA enhances luciferase production compared to the same dose composing of only luciferase mRNA although in a cell type specific manner. Toxicity caused by transfection of unmodified mRNA is mitigated with the delivery of NS1 mRNA and is observed only in NS1 without cleavage and polyadenylation specificity factor 30 kda (CPSF30) inhibition function. Conversely, delivery of mRNA encoding NS1 with CPSF30 inhibition function aggravated toxicity. Overall, we demonstrate that NS1 enhanced mRNA transfection through active evasion of innate immune responses and modulated cellular viability during mRNA transfection.
Introduction
I
While existing approaches that emphasize passive evasion from the innate immune system for the purpose of transfection enhancement are well developed, active immune evasion methods, however, are poorly investigated. The best known active immune evasion protein applied in mRNA delivery is B18R, a secreted protein from vaccinia virus that prevents IFN augmented innate immune responses by binding to IFN produced by mRNA transfected cells and sequester them from IFN receptors on cell membrane [18]. B18R protein is applied mainly as a media additive for in vitro mRNA transfections, especially in reprogramming applications [19]. Besides B18R, additional active immune evasion genes derived from the vaccinia virus have also been reported to be effective in mitigating toxicity of unmodified mRNA [20] and improving translation efficiency of self-replicating RNA [21]. Our group has also recently reported mRNA translation enhancement by co-delivering mRNA encoding nonstructural protein 1 (NS1), an active immune evasion gene derived from influenza A virus (IAV) [22]. Luciferase translation was exponentially enhanced by NS1 not only through innate immune activation/effector pathways (by inhibiting PKR and interferon regulatory factor 3 [IRF3]) but also through nonimmune regulatory pathway (by inhibiting cleavage and polyadenylation specificity factor 30 kda [CPSF30]).
Indeed, the accumulating evidence reported by the abovementioned studies supports the notion that mRNA therapeutics could benefit from active immune evasion strategies through the utilization of nature inspired viral immune evasion proteins. In this study, we further characterize mRNA translation enhancement brought by NS1. We show that higher mRNA translation was achieved in four different IFN competent cell types as rapidly as 6 h post transfection when reporter mRNA (encoding green fluorescent protein [GFP]) was co-delivered with mRNA encoding NS1. Cells transfected with NS1 mRNA produce less IFN, positively correlating with translation enhancement. There was no opportunity cost of delivering NS1 mRNA although in a cell type specific manner. Importantly, co-delivery of mRNA encoding NS1 protein also modulates toxicity induced by mRNA transfections. In particular, NS1 without the ability to inhibit CPSF30 enhanced viability of transfected cells while NS1 with the ability to inhibit CPSF30 moderately increased the toxicity of transfected cells. In conclusion, our results support an emerging concept of co-delivering mRNA encoding IAV derived NS1 protein that modulates protein expression and transfection toxicity.
Materials and Methods
Cells and reagents
Human foreskin fibroblast cells (BJ fibroblasts), murine macrophage cells (RAW 264.7), primary mouse embryonic fibroblasts (pMEFs), African green monkey kidney epithelial cells (Vero), human glioblastoma cells (C16), murine fibroblast cells (L929), and human cervix carcinoma cells (HeLa) were cultured in Dulbecco's modified Eagle's medium (DMEM) (high glucose) growth medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin–streptomycin. Human hepatocellular carcinoma cells (HepG2) were cultured in minimum essential medium (MEM) growth medium (with Eagle's basic salt solution) with the same supplements. All cells were from the American Type Culture Center, except for pMEFs (Lonza), and were cultured at 37°C in a saturated humidity atmosphere with 5% CO2. DMEM (high glucose), MEM growth medium (with Eagle's basic salt solution), penicillin–streptomycin 100 × solution, FBS, and trypsin 2.5% 10 × solution were purchased from Hyclone, GE Healthcare Life Sciences. Resazurin stock solution was prepared by dissolving 1 g of resazurin sodium salt (MP Biomedicals) in 100 mL sterile phosphate-buffered saline (PBS) and filtered through a 0.22 μm filter. Resazurin working solution (Alamar Blue working reagent) was freshly prepared before each experiment by diluting the stock solution with growth medium at a ratio of 1:250. The Alamar Blue reagent applied at this ratio was tested on BJ fibroblasts and determined to vary linearly with cell number. Stemfect RNA Transfection Kit (Cat# 00-0069) was purchased from Stemgent and used according to the manufacturer's protocol. Steady-Glo luciferase reagent and Glo lysis buffer were purchased from Promega.
Cloning of NS1 genes
A pCAGGS plasmid containing an open reading frame of NS1 of A/Hong Kong/156/1997 (HK97) was a kind gift from Adolfo Garcia-Sastre [23]. The open reading frame of HK97 was amplified by PCR (primers 5′ GGGGCGGCCGCTCAAACTTCTGACCTAATT 3′ and 5′ GGGGTCGACGCCACCATGGATCCAAACACTGT 3′) and cloned between SaI-I and Not-I sites of a pGEM4Z-A64 vector containing a T7 promoter and poly(A) tail (A64) creating a new mRNA transcription template pGEM4Z-HK97-A64 as reported in our previous work [22]. Cloning was verified by restriction digestion and sequencing.
In vitro transcription
In vitro transcription was performed as previously described [24,25]. Briefly, pGEM4Z-HK97-A64 was used as template for in vitro transcription using T7 High Yield RNA Synthesis Kit (Cat# E2040 NEB) in the presence of anti-reverse cap analog (Cat# S1411, NEB) according to manufacturer's protocol with a capping efficiency of ∼80% (based on 4:1 ratio of anti-reverse cap analog to guanosine-5′-triphosphate). IVT mRNA was purified with RNeasy Kit (Qiagen), quantified by spectrophotometry, and analyzed by agarose gel electrophoresis to confirm the synthesis of full-length mRNA.
Quantification of GFP mRNA expression
All mRNA transfections in this study were performed with Stemfect Transfection Kit according to manufacturer's protocol. For the analysis of GFP mRNA expression, cells were seeded overnight on 24-well plates at a density of 6 × 104 cells/well. Transfection was performed with 0.25μg/well of GFP mRNA, together with 0.25 μg/well of either NS1 mRNA or luciferase mRNA (control). The total dose was 0.5 μg/well. Media was changed and replaced with fresh growth medium 4 h post transfection. After another 2 h, cells were trypsinized, fixed with 1% paraformaldehyde, and GFP expression was analyzed with flow cytometry. Results represented data averaged from three independent repeats (mean ± SEM, n = 3).
Viability assay
For single transfection, cells were seeded overnight on 48-well plates at a density of 3 × 104 cells/well. Transfection was performed with 0.18 μg/well of either NS1 mRNA or luciferase mRNA (control), and 24 h later, media was replaced with Alamar Blue working reagent. After incubating for 2 h, fluorescence signals of the supernatants were measured using 544 nm excitation and 590 nm emission filter settings on a BMG LABTECH FLUOstar OPTIMA spectrophotometer. Blank wells without cells were used for background subtraction. Cells were then washed with PBS, added with fresh growth media, and incubated for another 22 h before the second Alamar Blue assay was repeated as described above. Results represented data averaged from four independent repeats (mean ± SEM, n = 4).
For serial transfection, cells were seeded overnight on 48-well plates at a density of 3 × 104 cells/well. Viability was tested with Alamar Blue working reagent as described above 4 h before the first transfection. Cells were transfected with 0.12 μg/well. Four hours post transfection, media was replaced with fresh growth media. After incubating for 2 h, viability was measured as described above. From day 2 onward (24 h after the first transfection), transfection with 0.12 μg/well, removal of particles, and viability assay were repeated as described for day 1 every 24 h. Results represented data averaged from four independent repeats (mean ± SEM, n = 4) and normalized to respective viability data obtained before the first transfection.
Quantification of luciferase mRNA expression
For zero-sum dosing experiments, cells were seeded overnight at a density of 3 × 104 cells/well on 48-well plates. Transfection was performed with a total dose of 0.12 μg/well of mRNA, consisting of a varying ratio of luciferase and NS1 mRNA. After 18 h, medium was aspirated, and Alamar Blue working reagent was added to each well to measure the cell viability. After this, cells were washed with PBS, lysed with 110 μL of Glo-lysis buffer, and subjected to 3 freeze–thaw cycles. Fifty microliters of cell lysate was then transferred to a white opaque plate (Nunc) and mixed with 50 μL of Steady-Glo Luciferase substrate (Promega). Bioluminescence was measured by BMG LABTECH FLUOstar OPTIMA spectrophotometer. Results represented data averaged from three independent repeats (mean ± SEM, n = 3).
Detection of IFN beta by enzyme-linked immunosorbent assay
Cells were seeded overnight at a density of 3 × 104 cells/well on 48-well plates. Transfection was performed with 0.2 μg/well of mRNA, and 18 h later, supernatant was collected for subsequent enzyme-linked immunosorbent assay (ELISA). Human IFN beta (IFN-β) in the collected supernatant was detected by sandwich ELISA using a capture antibody, rabbit polyclonal to IFN-β-C-terminal (Cat# ab186669), and a detection antibody, rabbit polyclonal to IFN-β (Biotin) (Cat# ab84258), both purchased from Abcam. Mouse IFN-β in the collected supernatant was detected with VeriKine Mouse IFN Beta ELISA Kit from PBL Assay Science according to the manufacturer's protocol.
Statistical analysis
Results are presented as mean ± SEM for the individual experiments. Comparisons between the groups were analyzed using one-way ANOVA, followed by multiple comparisons (experiment vs. control) using Student's t-test with GraphPad Prism. P < 0.05 was considered as statistically significant.
Results
Co-transfection with NS1 mRNA can establish rapid mRNA transfection enhancement in IFN competent cells
NS1 mRNA was co-delivered with GFP mRNA into fibroblasts (BJ fibroblasts), hepatocytes (HepG2), macrophages (RAW 264.7), primary fibroblasts (pMEF), and Vero cells. Meanwhile, luciferase mRNA (as a control) was co-transfected with GFP mRNA. As shown in Fig. 1A, we achieved high-baseline transfection efficiency based on the percentage of GFP-positive cells in all five cell types. Enhanced GFP mRNA expression was observed only in cells co-transfected with NS1 mRNAs, through greater right shifts of histograms, 6 h post addition of mRNA nanoparticles in all cell types except for Vero (Fig. 1B). The percentage enhancement, in terms of mean fluorescence intensity of GFP+ population (Fig. 1C), ranged from 29% (RAW 264.7) to 72% (pMEF). There was, however, no measurable increase in the percentage of GFP-positive cells (Fig. 1A). Our results showed that NS1 is capable of rapidly asserting its effects to enhance mRNA transfection in a wide range of cell types within a very short duration of time.

GFP mRNA expression 6 h after co-transfection with mRNA encoding NS1 or luciferase in five different cell lines. Cells seeded on 24-well plates were transfected with 0.25 μg of GFP mRNA and 0.25 μg of either NS1 or luciferase mRNA (control). Cells were then assayed 6 h after transfection.
Cells transfected with NS1 mRNA produce less IFN
Superior translational capacity and reduced cytotoxicity of IVT mRNAs are known to correlate with reduced activation of innate immune pathways [8]. Hence, we investigated whether there would be similar reductions in IFN production with NS1 mRNA. For this, we transfected all the four IFN-competent cells with either NS1 mRNA or luciferase mRNA (control) and measured secreted IFN-β in culture supernatants 18 h later by ELISA. As shown in Fig. 2A, there were statistically significant differences in secreted IFN-β levels between NS1 mRNA and luciferase mRNA in all four transfected cell types. In addition, the ELISA results for BJ fibroblast were further confirmed in BJ fibroblast IFN-stimulated response element (ISRE) reporter cells, which express luciferase in response to IFN. The result suggested that IFN pathways were activated to a lesser extent in NS1 transfected cells (Fig. 2B).
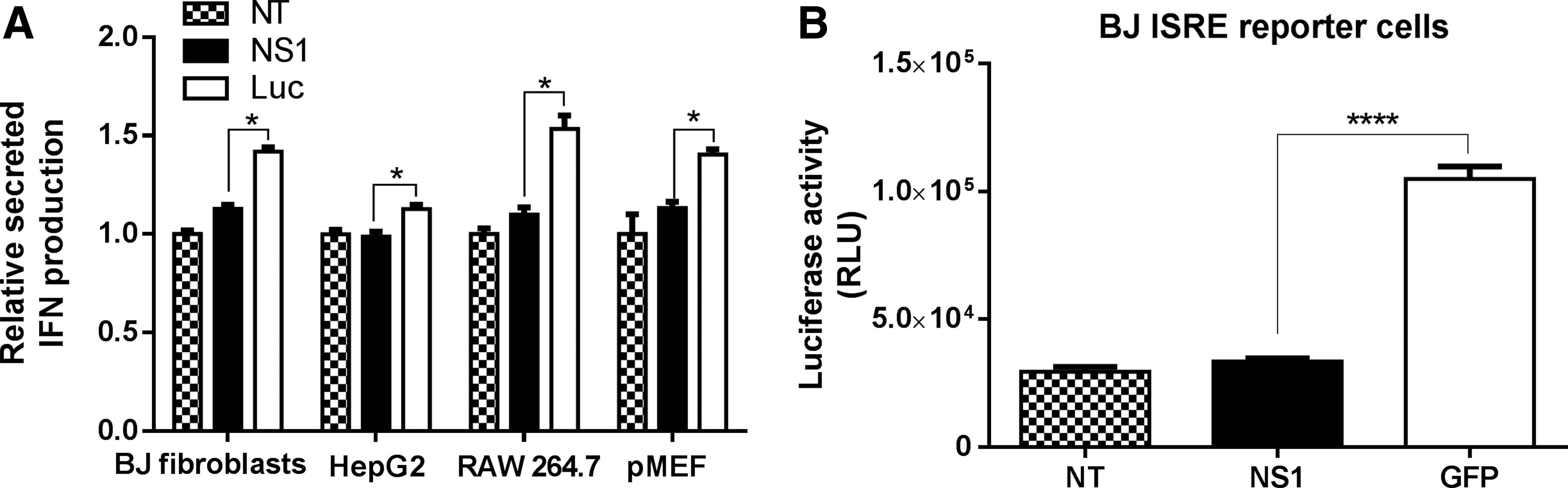
No opportunity costs of NS1 mRNA administration in certain cell lines
Since transfection enhancement was achieved by NS1 co-delivered in mRNA format, we questioned whether this co-delivery method would incur “opportunity costs.” In other words, whether substituting a small portion of luciferase mRNA with NS1 mRNA during mRNA transfection would affect luciferase production compared to a full unsubstituted dose composing only of luciferase mRNA. To answer this question, we transfected the four aforementioned IFN competent cell types with varying ratios of luciferase and NS1 mRNAs while keeping the total dosage constant at 0.12 μg. Luciferase expression was quantified 18 h post addition of the particles, which we observed was the time point when luciferase expression was the highest (data not shown). As shown in Fig. 3A, B, for BJ fibroblasts and HepG2, higher luciferase expressions were achieved in the groups transfected with 0.09 μg of luciferase mRNA and 0.03 μg of NS1 mRNA (ratio 3:1), compared to the positive control group which was transfected with an unsubstituted luciferase mRNA dose of 0.12 μg. The average enhancement observed in BJ fibroblasts and HepG2 was 13% and 47%, respectively. However, no similar observations could be made in RAW 264.7 and pMEF cells (Fig. 3C, D) as luciferase expression could not exceed that of cells transfected with 0.12 μg of luciferase mRNA.
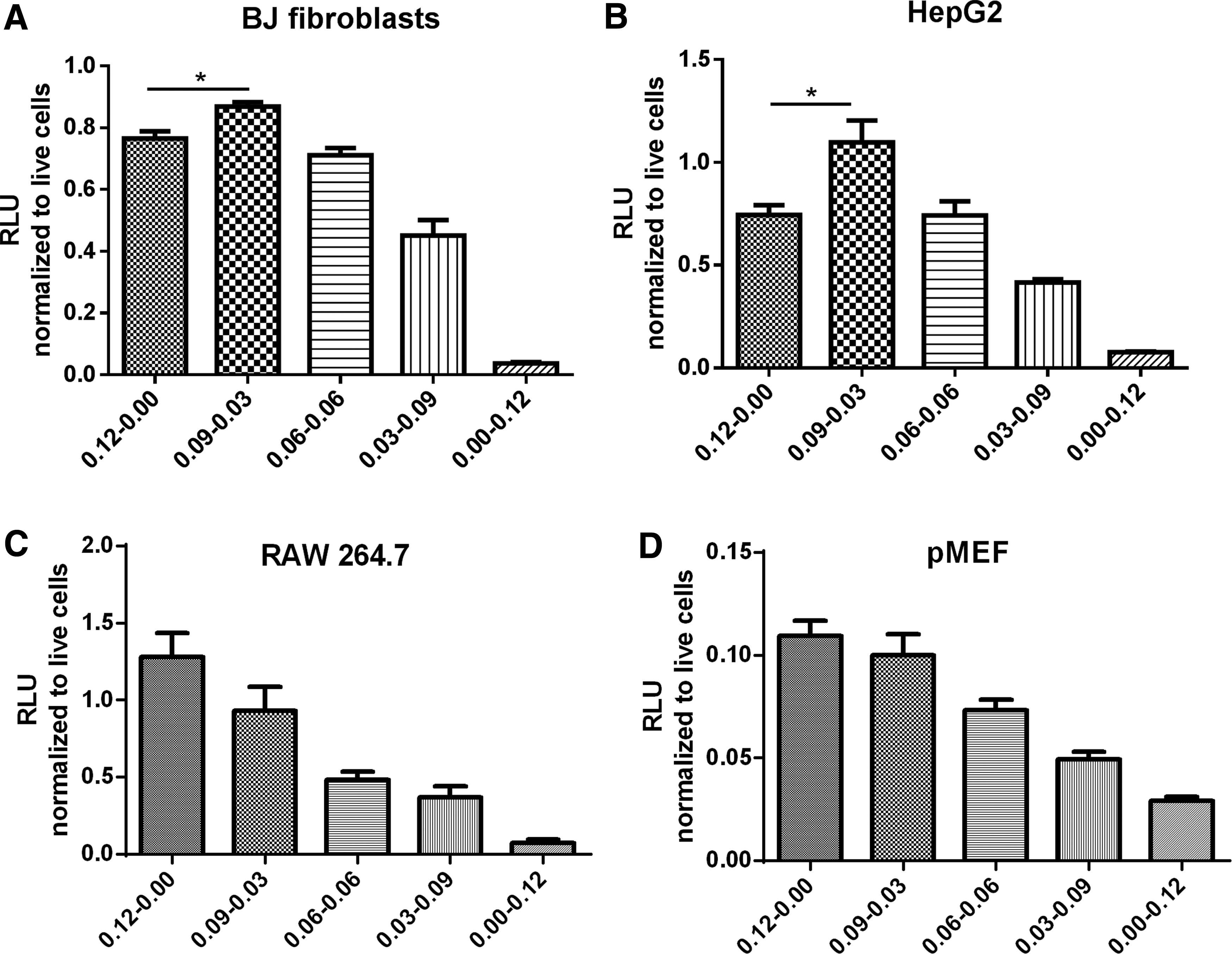
Luciferase expression in
RNA-induced toxicity is mitigated by NS1 without CPSF30 inhibition function
After confirming mRNA transfection enhancement by co-delivery of NS1 mRNA, we further investigated whether NS1 protein expressed from mRNA could mediate cytotoxicity. To address this question, we first transfected all five aforementioned cell types with mRNA encoding NS1 or luciferase (control) and monitored cell viabilities 24 and 48 h post transfection. As shown in Fig. 4, NS1 mRNA did not increase cytotoxicity in the IFN competent cells, compared to luciferase mRNA. Interestingly, a slight increase of viability was consistently observed in all NS1 groups, indicating that the toxicity caused by mRNA transfection could be mitigated by the expression of NS1. To elucidate NS1's capacity to mitigate mRNA induced toxicity, we performed a serial mRNA transfection on BJ fibroblast through daily dosing for five consecutive days and found that NS1 was indeed able to reduce the toxicity during serial mRNA transfection (Fig. 4E).
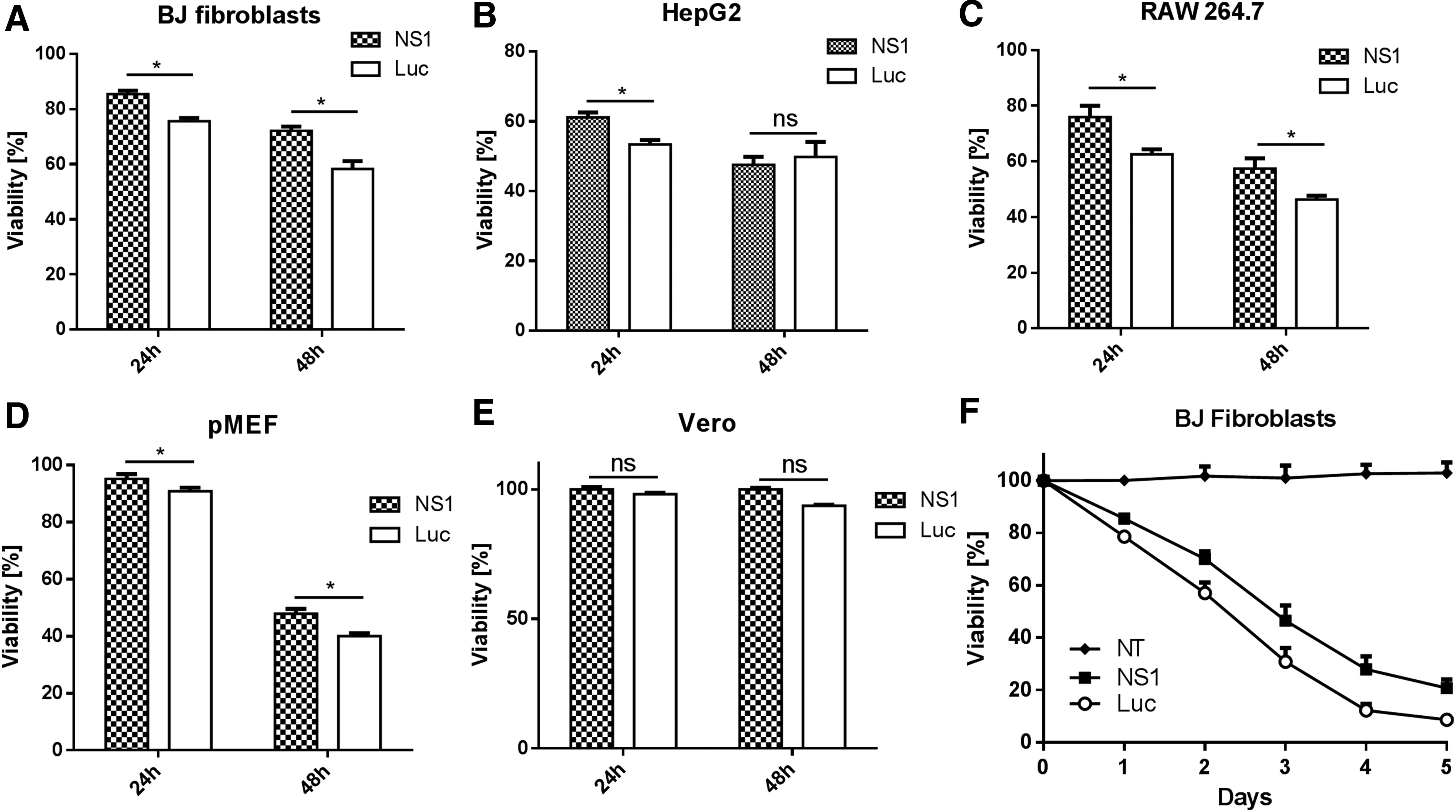
Viability of mammalian cells 24 and 48 h after transfection with NS1 or luciferase mRNA. mRNA as indicated was transfected into
We further investigated toxicity effects of two different types of NS1 (Table 1): the first type does not inhibit the CPSF30, while the second type inhibits CPSF30 (Table 1). The NS1 mRNA evaluated in this study belongs to first type and is derived from A/Hong Kong/156/1997 strain (HK97) (Table 1). As shown in Fig. 5, cells transfected with mRNA encoding non-CPSF30 inhibiting NS1 (HK97, PR8, SH, and CAL) were consistently more viable than GFP control at all tested doses. In contrast, cells transfected with mRNA encoding CPSF30 inhibiting NS1 (VN, TX91, SHM, PR8S) were consistently less viable than GFP control at all tested doses. Notably, this trend was consistently observed in four different cell types.

Cytotoxicity evaluation of NS1 with and without CPSF30 inhibition function in various cell types.
NS1, nonstructural protein 1; CPSF, cleavage and polyadenylation specificity factor.
Discussion
Despite the benefits of utilizing IVT mRNA to induce the expression of therapeutic proteins, innate immune responses triggered during the delivery of mRNA remains a lingering problem. Consequently, much effort has been made to reduce the immunogenicity of IVT mRNA to enhance its translation efficiency for nonvaccine applications. IAV contains a segmented genome, which consists of eight gene segments that encode 11 proteins, including NS1 [26]. NS1, which is a highly conserved protein with an average size of ∼230 amino acids, is encoded by the eighth segment of IAV's genome and abundantly produced during IAV infection.
There are two aspects of NS1 that makes it a novel immune evasion protein in the context of mRNA delivery. First, NS1 is a multifunctional protein. This property is imparted by the nature of RNA viruses, which tend to encode multifunctional proteins as an evolutionary adaption to their small genomes. In contrast, larger genome sizes of DNA viruses allow a greater number of genes to be devoted to host control [27] and consequently, immune evasion functions are spread among monofunctional proteins encoded by multiple genes. For instance, the vaccinia virus (DNA virus) has ∼250 genes, and more than 20 genes have been identified to be associated with immune evasion. While some function extracellularly to bind with IFN-α/β (ie, B18R) or IFN-γ (ie, B8R), others function intracellularly to inhibit IFN pathways, by suppressing the IFN-induced proteins like PKR (ie, E3L) or inhibiting the activation of IRF3 (ie, N1L) [28]. Co-delivering monofunctional immune evasion genes may not be sufficiently efficient to counteract multiple innate immune pathways triggered by mRNA. Second, NS1 is dedicated to immune evasion functions. For many RNA viruses, immune evasion functions are performed by multifunctional proteins in charge of maintaining viral replication of the virus, which are irrelevant in nonviral IVT mRNA delivery. For example, NS2B3 of the dengue virus can inhibit IFN production by disrupting the phosphorylation of IRF3. Meanwhile, it is also a protease complex responsible for cleaving the polyprotein translated by the positive-sense RNA genome [29]. In another example, NS34A protease of the hepatitis C virus is a complex consisting of NS3 and its cofactor NS4A, which is crucial for viral RNA replication and virion formation. Meanwhile, it can also block TLR3 signaling by cleaving the signaling protein TIR-domain-containing adaptor-inducing IFN-β (TRIF) [30]. Taken together, NS1 is novel because it is a protein that possesses multiple functions, which are exclusively involved in immune evasion activities. NS1 can counteract host cell innate immune response by suppressing the activity of IRF3 [31] or by obstructing the overall gene expression in the host cell by suppressing polyadenylation of pre-mRNAs in the nucleus [32]. Furthermore, it has also been reported to inhibit activities of IFN-induced proteins such as PKR [33] and (2′-5′-oligoadenylate synthetase (OAS) [34].
The best approach to apply NS1 is to co-transfect NS1 mRNA with mRNA encoding the gene of interest. Unlike B18R, NS1 works inside the cells and is therefore preferably delivered in nucleic acid form to take advantage of nonlinear amplification of NS1 from the protein translation process. The reason to choose mRNA but not plasmid as the nucleic acid format is because mRNA transfects more quickly (faster kinetics) and efficiently (more cells transfected because mRNA works in the cytoplasm), allowing NS1 to take effect as soon as possible. While it is favorable to pre-establish NS1 presence within host cell before mRNA transfection, co-transfection instead of pretransfecting cells with NS1 mRNA is no doubt more practical. Moreover, pretransfecting cells with NS1 mRNA may backfire as it would hypersensitize the cell's innate immune system and compromise the subsequent transfections. In addition, it is also a more sensible imitation of the natural working mechanism of NS1, which is co-expressed with viral genes in the host cells to provide for immune evasion after the entry of the virus into the cytoplasm.
In this study, we tested the efficacy of NS1 in five different cell types that represent various mRNA delivery applications. Human skin fibroblasts (BJ fibroblasts) were selected for their high relevance in cellular reprogramming [9]. HepG2 were chosen because the liver is an attractive target organ for nonviral gene therapy. Macrophages (RAW 264.7) were selected to represent immune cells [22]. Primary mouse embryonic fibroblasts (pMEFs) were included for their higher biological authenticity and relevance to in vivo application. Finally, Vero cells were used as control because they do not produce IFN, the main innate immune response induced by unmodified mRNA. Co-transfection of NS1 and GFP mRNA led to rapid GFP expression enhancement in every IFN competent cell type (Fig. 1), suggesting a wide host range property and fast kinetics of NS1. In contrast, no enhancement was observed in Vero cells, an IFN-deficient cell type, indicating that NS1-mediated enhancement is likely to be IFN related.
Indeed, NS1 was found to mitigate innate immune response brought by mRNA transfection (Fig. 2). IFN-β, a surrogate of innate immune response, was detected in the supernatant of the transfected cells by ELISA at significantly lower levels in all cell types transfected by NS1 mRNA. The lower luciferase production in NS1-transfected BJ ISRE reporter cells further confirmed that NS1 was able to mitigate the activation of IFN-stimulated JAK/STAT pathway. Altogether, our results suggest that NS1 boosts mRNA transfection through counteracting type I IFN. While transfection enhancements were evaluated by comparing NS1/reporter gene with control gene/reporter gene mRNA formulations (Fig. 1), a more practical comparison would be between to compare NS1/reporter gene and a full dose of reporter gene (ie, without control gene as a filler) as previously reported [22]. By substituting a fraction of luciferase mRNA with NS1 mRNA without increasing the total mRNA dosage, higher luciferase expression could be achieved (compared to an unsubstituted dose of luciferase mRNA) although only in BJ fibroblasts and HepG2 (Fig. 3). This suggested that while NS1 might be effective across different cell types (Fig. 1), its practical value would nevertheless be cell type dependent.
It is noted that the “NS1” applied in this article was derived from A/Hong Kong/156/1997 (ie, NS1-HK97) in contrast to NS1-TX91, which was applied in our pioneering work on NS1 for mRNA delivery. The difference between NS1-HK97 and NS1-TX91 is that the latter engenders CPSF30 inhibition function and, therefore, enhances mRNA translation significantly better than NS1-HK97. When we first started working with NS1, we had assumed that NS1 derived from different strains would have similar performance, and hence, cytotoxicity was our selection basis. At that point we were unaware of the impact of NS1's CPSF30 inhibiting function and its remarkable ability to enhance mRNA translation. Consequently, NS1-HK97 was selected based on preliminary toxicity screen, which revealed that cells transfected with NS1-HK97 not only had the highest viability but also they were more viable than nontransfected cells. After we have already generated a sizeable amount of data based on NS1-HK97, we stumbled upon the discovery that NS1 protein engendering CPSF30 inhibition function (exemplified by NS1-TX91) was unusually effective in enhancing mRNA translation and diverted our efforts to NS1-TX91 and characterized it against one cell type (BJ fibroblasts). In this article, we additionally provided evidence that correlates translation enhancement with IFN production (ELISA) in four different cell types, thereby corroborating the results on BJ fibroblasts from our previous article [22] although NS1-HK97 may not be the best for enhancing mRNA translation because it does not engender CPSF30 inhibition function.
A novel observation made in this study was that NS1 modulated cell toxicity induced by transfection of unmodified mRNA and was directly correlated to NS1's ability to inhibit the CPSF30 subunit. CPSF30 inhibition is a consensus immune evasion mechanism uniquely engendered by influenza A to counteract innate immune response of host cells during very early stages of viral infection [23]. While the detailed mechanism is reviewed elsewhere [35,36], the consequence of CPSF30 inhibition is the inhibition of host gene expression through reduction of poly A tail lengths of endogenous pre-mRNAs, thereby preventing their nuclear export and cytoplasmic stability. The consensus amino acid determinants in NS1's primary sequence for CPSF30 inhibition have been determined. NS1 will inhibit CPSF30 if its primary sequence contains all four consensus amino acids, namely 103F, 106M, 108K, and 189D (Table 1) [23]. Since different IAV strains give rise to NS1 with different primary amino acid sequences, NS1 derived from certain IAV strains that do not contain all four consensus amino acids would not inhibit CPSF30. During this study, which was conducted using mRNA encoding NS1 derived from A/Hong Kong/156/1997 that did not inhibit CPSF30 (Table 1), we repeatedly noticed a higher viability post transfection compared to controls (Fig. 4) and even to nontransfected controls (Fig. 5B). As we had several NS1 mRNAs in our hands, we screened cellular toxicity induced by the transfection of mRNAs encoding NS1 protein derived from different viruses (Table 1). A consistent trend emerged which showed that cells transfected with mRNA encoding NS1 without CPSF30 inhibition function were always more viable than those transfected with mRNA encoding GFP or NS1 with CPSF30 inhibition function (Fig. 5). This trend was further confirmed when the experiment was extended to other cell types, indicating that this trend was not a cell type specific effect. Indeed, it is known that NS1 possesses either antiapoptotic [37,38] or apoptotic [39] properties in a cell type specific way. However, the modulation of cell viability in mRNA transfected cells and its dependence on NS1's CPSF30 inhibition function has not been reported. In our previous study, we reported that co-delivery of NS1 engendering CPSF30 inhibition function exerted a remarkably positive effect on mRNA translation [22]. In this study, we show that such NS1 bears a moderately negative effect on cell viability, which can be considered tolerable (>80% viability) under normal transfection dose. Importantly, our study reveals that NS1 lacking CPSF30 inhibition function can enhance cell viability of mRNA transfected cells. A similar observation was also reported in another study using immune evasion genes derived from the vaccinia virus [20]. Taken together, it can be concluded that co-delivering NS1 mRNA offers advantages in terms of enhancing mRNA translation and reducing transfection toxicity.
In conclusion, we demonstrate that co-delivery of NS1 mRNA (1) improves mRNA translation with corresponding reduction in IFN production in a wide range of cell types (human fibroblasts, HepG2, RAW 264.7, pMEFs) and (2) modulates cell viability during mRNA transfection. Co-delivery of active immune evasion genes reveals an additional approach to mRNA delivery that can impact nonviral gene medicine and warrants further research.
Footnotes
Acknowledgments
This research was supported by MOE AcRF Tier 1 (R-279-000-430-133; R-279-000-471-112) and A*STAR SERC 2015 PSF grant (R-279-000-475-305), NMRC OFYIRG (R-279-000-492-511), and NUS research scholarship (L.Y.). The authors thank Dr. Adolfo Garcia-Sastre for providing the NS1 gene and Dr. L.J. for his assistance in cloning of NS1 genes.
Author Disclosure Statement
No competing financial interests exist.