
Abstract
Although recent clinical successes of antisense, splice-switching, and siRNA oligonucleotides have established the therapeutic utility of this novel class of medicines, the efficient systemic application for non-liver targets remains elusive. Exploitation of active receptor-mediated targeting followed by efficient and productive cellular uptake is required for enabling the therapy of extrahepatic diseases on the expressional level. Evasion of liver accumulation and organ-specific targeting and also efficient cytosolic delivery after endosomal internalization are currently insufficiently solved issues. Lipid and polymer-based nanoparticles can be engineered for efficient cellular uptake and enhancement of endosomal escape, but are characterized by preferential liver accumulation based on biodistribution largely determined by particle size and biophysical properties. Oligonucleotide bioconjugates with receptor-binding ligands have been evolved for highly efficient targeting, but frequently result in a large extent of endosomal entrapment and consequently a lack of sufficient cytosolic concentrations. Non-immunoglobulin protein-based receptor recognition affords high cell-type selectivity and is promising for achieving nonhepatic oligonucleotide targeting. The use of such novel protein scaffolds, including designed ankyrin repeat proteins (DARPins), for oligonucleotide delivery is attractive for achieving effective tissue targeting. Issues for further development and optimization to advance approaches for extrahepatic oligonucleotide delivery by nanoparticles or bioconjugates are discussed.
Delivery Approaches for Therapeutic Oligonucleotides
D
Patisiran and most other siRNAs in late-stage clinical trials are aiming at modulating genes in the liver, either using liposomal formulations or GalNAc bioconjugation. Current clinical and preclinical successes are largely restricted to local applications or to hepatic targets after systemic injection, due to an intrinsic preference for accumulation of respective agents in the liver. For different reasons, phosphorothioate antisense oligonucleotides [7], N-acetyl galactosamine (GalNAc) conjugates [8,9], and liposomally encapsulated siRNAs [10] all show strong hepatic targeting. This is in principle due to binding to plasma proteins (phosphorothioates), receptor-mediated hepatocyte uptake through the asialoglycoprotein receptor (GalNAc conjugates), and particle-size dependent enrichment in liver and other organs with larger fenestrations or pores in the endothelium (liposomes and other nanoparticles) [11].
In the case of nusinersen, which is applied locally through injection into the spinal canal, the effect on restoration of functional SMN protein is limited to the central nervous system [12,13]. Although significant benefit is achieved for the patients, a therapeutic effect in the periphery would arguably further enhance clinical outcomes [14]. A proportion of the injected dose of nusinersen is entering the blood circulation and is quickly eliminated as the drug lacks phosphorothioate linkages which are essential for plasma protein binding and consequent longer retention in the circulation [7]. The need of modulating splicing or silencing genes beyond the liver in a therapeutically relevant extent is insufficiently met by available technologies [11].
To completely fulfill the promise of becoming a third large therapeutic class besides small molecules and recombinant proteins, this issue remains to be solved. For avoiding the large extent of hepatic accumulation, actively targeted delivery approaches appear to be a necessity. A wealth of different strategies for targeted delivery of nucleic acids and other cargoes has been developed and evaluated, including immunoliposomes [10,15], other targeted nanoparticles [16–18], antibody-oligonucleotide conjugates [19], and other bioconjugates [20–23]. Despite important virtues, novel nonimmunoglobulin proteins have only rarely been adopted for oligonucleotide targeting. This article summarizes recent developments of using these protein scaffolds for extrahepatic targeting of therapeutic oligonucleotides.
The Right Choice of Targeting Ligands Is Essential
It is self-evident that successful drug targeting depends heavily on used ligands, which in most cases target receptors that are more or less unique for a specific organ or tissue. Optimal targeting strategies need to take the receptor abundance into account, but also its uptake route and kinetics. In terms of the respective ligand, high-binding specificity and affinity and facile attachment directly to the oligonucleotide cargo or the delivery system have to be considered, as well as general drug development aspects, including lack of toxicity and immunogenicity, economical scale-up and GMP production, and impurity profiling [24,25]. Critical parameters and optimization aspects differ greatly for covalent oligonucleotide-ligand conjugates on the one hand and targeted nanoparticle systems on the other hand. These include the influence of receptor affinity, the choice of attachment site, and accessibility after tethering to the cargo or a particle.
Both target receptor and the respective ligand of the delivery system need to fulfill several requirements as follows: The receptor should have strong expression in the targeted organ/tissue and limited abundance in other regions [26]. In addition, it needs to be accessible for binding of the delivery system and result in cellular uptake upon binding [27]. The uptake kinetic is particularly important so that the bound delivery system is able to reach endosomes and finally the cytosol in sufficient concentrations [28].
The final and arguably the most difficult barrier to overcome is the endosome. Without any endosomal escape enhancing mechanism, only insufficient cytosolic concentrations will generally be reached [29]—a sole exception being the asialoglycoprotein receptor, which rapidly internalizes large quantities of GalNAc-modified oligonucleotides so that even a low percentage of escaping cargo is sufficient [30]. Proteins, peptides, aptamers, and small molecules all can be suitable targeting moieties, depending on the particular receptor and the availability of a corresponding binding site [31,32], but nonantibody protein scaffolds generally have better affinity and enable more specific target recognition.
Protein–protein interactions are characterized by high specificity and binding affinity, and antibodies remain to be nature's gold standard for highly specific protein recognition [25,33,34]. However, their complex multidomain structure, their large size, and their reliance on correct posttranslational modifications, which is indispensable for high specificity and affinity, negatively impact stability during production, isolation, and processing. Derivatization for drug targeting is particularly complicated, because a unique chemical functionality for linker attachment is lacking, and generally, a mixture of products with different number of tethers and attachment sites is produced [35].
Antibodies and their derivatives have been developed for specific delivery of small molecules for many years. Although antibody-drug conjugates have improved the therapy of some cancers and despite recent successes in research and development, overall the therapeutic development has been long and complicated [36]. Antibody fragments are smaller, and for targeting purposes, Fab and single-chain Fv (scFv) fragments, both lacking the immune activating Fc region, are attractive [37]. Initial therapeutic development of extrahepatic oligonucleotide targeting with proteins has been achieved with single-chain Fv fragments (scFv) [38,39]. Since then, other protein scaffolds with simpler structures have come into focus for oligonucleotide delivery.
During the last 20 years, several next-generation protein scaffolds have been discovered and engineered for stability, specificity, and affinity [40] (Fig. 1). Among those are affibodies [41], anticalins [42], and designed ankyrin repeat proteins (DARPins) [43,44]. Besides their potential therapeutic utility per se, novel protein binder scaffolds are particularly attractive for targeting purposes, because of their much more facile generation, production, and derivatization [40,45,46]. Within these protein classes, binders against desired purified antigens or receptors can generally be developed by producing libraries and phage or ribosome display technologies [47,48]. For oligonucleotide targeting, only affibodies and DARPins have so far been reported.
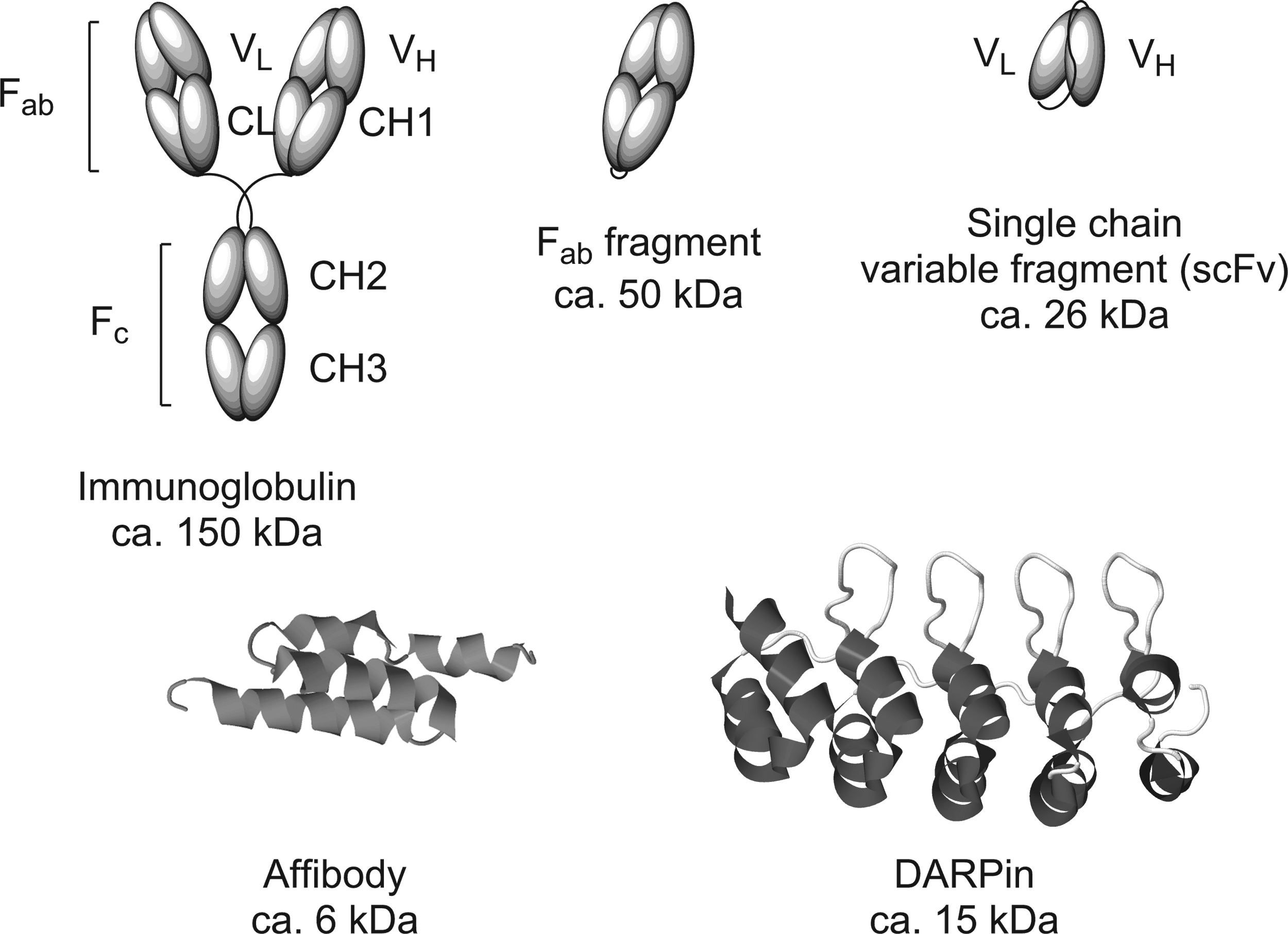
Schematic illustration of a monoclonal antibody and respective fragments for therapeutic targeting and cartoon representations of an affibody and a DARPin. Antibodies and their fragments have excellent protein binding specificity and affinity, but their structure and partially the large molecular size complicate the use for drug targeting. Non-immunoglobulin binding proteins can be evolved to a similar extent of specific and high-affinity binding, but allow facile derivatization for use for receptor-mediated oligonucleotide targeting. DARPin, designed ankyrin repeat protein.
Oligonucleotide Delivery with Affibodies
The design of affibodies is based on the immunoglobulin binding domain of protein A and consists of three bundled alpha helices [49]. A fusion protein of an RNA binding domain and an ErbB2 specific affibody was successfully used for targeting siRNA to Her2-expressing tumor cells and was effective in suppressing in vivo tumor growth in a xenograft mouse model [50]. This study used a noncovalent noncharge-based complexation of siRNA to the dsRNA binding domain of the engineered protein, and the affibody domain ensured rather specific, yet apparently moderate cellular uptake. In a xenograft model, the fusion protein was able to deliver siRNA to the tumor followed by silencing of the target genes.
With a nanoparticle derived from HBV L protein and surface antigen and lipids, the engraftment of a Her2-selective affibody to these particles resulted in higher GFP silencing in Her2-positive cell lines in vitro [51]. While the affibody-receptor interaction afforded superior uptake in Her2-positive cells and the lipids destabilized endosomes, the complexity of the particles complicates further development.
Designed Ankyrin Repeat Proteins for Oligonucleotide Delivery
DARPins are composed of ankyrin repeat motifs, the natural counterparts of which consist of usually 33 amino acids and are components of ankyrin repeat proteins [52]. The rationally designed consensus DARPin consists of N- and C-capping ankyrin repeat motifs, which have been developed for high stability and for correct folding in bacterial expression systems. Usually three internal repeat motifs with a number of variable amino acids each are present in each DARPin, which generates an extended target interaction interface essential for protein–protein binding [44].
The structure of repeat proteins allows for modification or exchange of individual repeat units without alteration of the tertiary protein structure. DARPins have been engineered for stability, while retaining their initial high-binding affinity and specificity. The lack of posttranslational modifications allows quick and high-yield expression from bacterial hosts, making them easily and economically available in sufficient quantities. Binders with affinities down to the picomolar range against a broad array of targets can be selected from libraries using ribosome display [44,48].
Ribosome display is an entirely in vitro method, and the lack of a bacterial transformation step, necessary for phage display, avoids the potential loss of library diversity [53]. Proteins isolated from each round may be further modified through error-prone PCR or directed mutations [54]. One issue for successful ribosome display is an efficient reverse transcription reaction of the isolated RNA and subsequent cDNA amplification and the need to strictly avoid nucleases. DARPins, with their advantageous folding behavior, are particularly suited for cell-free translation, and binders against many targets have been developed, including EpCAM [55], ERK [56], JNK [57], Her2 [58], IgE-receptor [59], HIV-gp120 [60], and many others.
In clinical development, an anti-VEGF-A DARPin was evaluated for treatment of age-dependent macular degeneration in phase I/II trials, showing good safety and preliminary efficacy, and a phase III trial is ongoing [61]. Owing to the lack of internal cysteines, DARPins can easily be derivatized for generating unique binding sites for linker attachment through thiol-reactive chemistry [62]. In addition, metabolic engineering has been used for site-specific incorporation of an azido-modified amino acid, which can subsequently be used for ligand or linker attachment through strain-promoted alkyne-azide cycloaddition (SPAAC).
Exploiting these modular characteristics and an orthogonal conjugation strategy, a DARPin-monomethyl auristatin F (MMAF) conjugate with additional albumin attachment for half-life extension has been generated [63]. Compared to the free cytostatic drug, an increase in cytotoxicity for antigen-positive cell lines was found and a decrease in antigen-negative cells. This example illustrates the facile assembly of DARPin bioconjugates and the possibility for rational design of chemically defined molecules.
Are Nanoparticles or Bioconjugates the Superior Choice for siRNA Delivery?
An essential issue is whether nanoparticles [64] or chemical bioconjugates [32] are used for oligonucleotide receptor targeting [23]. Both have their respective inherent advantages and drawbacks. Liposomes and other nanoparticles are effectively shielding the encapsulated or complexed nucleic acid cargo from enzymatic degradation and preventing renal filtration and rapid elimination [64]. Methods are available for grafting targeting ligands by either assembling the individual components, one of which includes the ligand, or by chemical attachment of the ligand to the prebuilt nanoparticle [65,66].
It is generally complicated to steer the number of ligands per particle precisely and often a complex mixture of particles with differing number of targeting molecules results. Since tens to hundreds of proteins or other targeting molecules are often found on the particle surface, the binding to the target cell is heavily influenced by concurrent binding of several molecules [67–69]. Consequently an avidity effect results, meaning that individual protein binding affinity is of minor importance, and also lower affinity molecules result in adequate association of particles to the target cells [70]. However, this effect increases homing to cells with only low-target receptor abundance and can also affect off-target binding [71].
It is important that the targeting molecule is available for protein interactions, and thus, its attachment on the outer particle layer and prevention of excessive absorption of plasma proteins (making up a biocorona) are essential [72]. To improve particle properties, the surface can be modified, for example, by PEGylation or by preabsorption of proteins [73]. In contrast, the biodistribution of particles is to a great extent determined by their size and biophysical characteristics [74]. Importantly, particles tend to accumulate in tissues with larger endothelial pores or fenestrations, which are mainly liver, spleen, and some tumors [75]. Thus, nanoparticle targeting approaches may fall short, especially when a cell-surface protein of tissue cells is the target; the nanoparticle might never come close to its target receptor localized in organs with tight endothelial barriers.
In research publications, targeted particles are often described as successful if the accumulation within the target organ is increased at least 1.5 or 2-fold compared to a nontargeted system, in the case of the brain or other tissues that are difficult to reach, that corresponds to well below 2% or even 1% of the total dose [23,76,77]. While such results may constitute an important progress in basic research, a direct translation to therapeutic use is questionable. Furthermore, particles with sizes over 100 nm are prone to be intercepted by the mononuclear phagocyte system (MPS, also known as reticuloendothelial system RES) [78], the extent of which increases with size, and due to the biocorona, in vivo particle sizes may frequently be larger than those measured in vitro.
DARPins can be relatively easily attached to nanoparticles by standard conjugation chemistries. Recently, SPAAC was used to tether an azido-modified DARPin to the surface of cyclooctyne-functionalized liposomes [69]. While the reaction to fully assembled liposomes proceeded much less efficiently and more slowly compared to a small molecule, a certain guidance toward the number of attached DARPins was achieved using variations of protein:liposome reaction ratios. It was shown that increasing the number of DARPins beyond a threshold of only a few molecules per particle failed to increase in vitro association to target positive cells [69]. Although DARPin functionalized liposomes have not yet been used for oligonucleotide delivery, the system could be adapted for nucleic acid lipid particles.
Initial successful oligonucleotide targeting was afforded by complexes of oligonucleotides with fusion proteins consisting of an scFv and a basic peptide [38,39]. The production of highly charged fusion proteins was difficult, and purification from expression systems needed denaturing conditions with subsequent refolding [38]. We generated a similar construct with an EpCAM-targeted DARPin, which was able to transfect cells in vitro and had high specificity for EpCAM-positive cell lines [79]. Such systems allow for facile complexation by simple mixing of protein and oligonucleotide in appropriate buffers and can thus be quickly adopted for varying antisense or siRNA sequences [33]. The cationic component is likely contributing to efficient functional uptake by destabilizing endosomes after internalization, but on the other hand raises potential safety issues [80]. Intermolecular aggregation can be caused by densely charged fusion proteins, which results in generation of larger particles of up to 200 nm in hydrodynamic radius for DARPin fusion proteins (unpublished data).
Since the association of the polyanionic oligonucleotide and a peptide consisting of arginine and lysine amino acids is based on electrostatic interactions, the complexation is both dynamic and relatively weak. Reports of similar scFv fusion proteins reported high capacity and indicated absence of significant aggregation tendency; these properties are apparently influenced by the structure and size of the binding domain, and the small size of DARPins seems to hamper the development of complexes. Upon preparing complexes even with low surplus of nucleic acid cargoes, a certain proportion is left uncomplexed. Although the particle size is sufficient for long circulation times, eventually dissociated oligonucleotides are eliminated rather quickly. Because of these issues, binding to the selected antigen and target cells needs to be proven after final assembly, to exclude that the availability of the targeting protein is compromised.
Taken together, these characteristics complicate reproducible, efficient, and safe in vivo use and also cause potentially serious manufacturing and stability issues. Thus, the development of fusion protein oligonucleotide complexes has not been taken much further toward clinical use since the concept had been reported for the first time more than 10 years ago. Further options for improving on this concept are the use of additives, in particular for enhancing loading capacity and maintaining stable complexation, as well as agents that increase endosomal escape [33].
In the case of nanoparticles, a strategy for extravasation may be required. One possibility is to select ligands for targets present at the endothelium, but instead of intracellular uptake and eventual re-transport to the luminal side, transcytosis needs to be triggered [81,82]. The mainly exploited receptor-ligand pair for transcytosis is the iron-binding glycoprotein transferrin and its respective receptor CD71 [83]. The transferrin receptor has the advantage of being recycled to the cell surface together with the iron-less apotransferrin, instead of the receptor and ligand being degraded in lysosomes. The rapid turnover of this system makes it attractive for targeting approaches. However, its selectivity is limited, as the receptor as an essential part of the iron metabolism is found in endothelia of nearly all tissues.
Recently, a siRNA covalently attached to an anti-CD71 Fab fragment was shown to induce gene silencing in skeletal and heart muscle in mice [84]. Assessed by silencing of the target genes, HPRT and myostatin, the effect was stronger and more prolonged than in the liver, reaching a plateau after 3–7 days in muscle, while peaking after 24 h in the liver. Transferrin-modified lipoplexes [85] and polyethylenimine particles [86] have also been used for in vitro siRNA uptake into neuronal and tumor cells, respectively.
Being single macromolecules, targeted oligonucleotide bioconjugates have largely different characteristics compared to particles [20,87]. The lack of being shielded from nucleases mediates the need for chemical derivatization to prevent rapid degradation. Appropriate chemical modification patterns that inhibit nuclease cleavage but preserve pharmacological activity have been developed and optimized for siRNAs, as well as antisense and splice-switching agents [88]. The success of GalNAc conjugates in preclinical and clinical evaluations proves that the stability issue is largely overcome.
To avoid quick renal elimination, bioconjugates would need to have a minimum size of around 6–10 nm, corresponding to ca. 20–50 kDa [89]. However, extravasation is likewise more rapid for smaller molecules, and strong binding (and cellular uptake) to the target would counteract elimination [90]. Again, GalNAc conjugates are proof that also molecules below the renal filtration limit and without strong plasma protein binding are potential drugs. The elimination threshold is not reached by conjugates with very small protein ligands, but most targeting proteins together with the oligonucleotide cargo surpass the limit. Further structural amendments such as PEGylation can be considered to modulate circulation times and biodistribution [21].
Unlike lipid-based and other nanoparticles, bioconjugates are generally not associated with a biocorona, and thus, the protein receptor interaction is not interfered with. The binding affinity can be expected to be very similar to that of the protein ligand, as long as oligonucleotide attachment is afforded on a site that is not essential for receptor binding, the linkage does not cause any steric hindrance or conformational change of the protein, and the polyanionic charge of the oligonucleotide is not too close to the cell membrane to cause repulsion. For rational approaches and pharmaceutical development, site-specific attachment and thus generation of a structurally homogenous compound are highly recommended. Thus, the chemical properties of the protein targeting ligand are decisive, and novel scaffolds such as DARPins with their facile site-specific introduction of unique functionalities have essential advantages over antibodies and antibody fragments [33,35,40,44]. Attachment sites on both C-and N-termini do not interfere with protein binding, which is exerted by the internal repeat domains.
We have recently generated DARPin-siRNA conjugates, joined by disulfide, thiol-maleimide, or triazido (generated by SPAAC) linkages (article in preparation). These conjugates all bind with high affinity and specificity to target positive cell lines, in essentially the same extent as the unconjugated parent DARPin. Furthermore, the conjugates were all taken up only in receptor-positive cells in vitro, lacking any internalization in target-negative cell lines. However, in terms of functional delivery assessed by gene silencing, the disulfide linked conjugate resulted in superior gene knockdown despite lower overall uptake compared to the more stable thiol-maleimide and triazido linked compounds. Our data indicate that cellular internalization is afforded through an endosomal route, and endosomal escape is the limiting step. Cleavage of the oligonucleotide-protein link at some point during or after successful internalization seems to be beneficial. In this regard, the targeted receptor and its uptake kinetics appear to be key issues for successful extrahepatic targeting.
The clinically used asialoglycoprotein receptor for GalNAc conjugate uptake profits from unique properties. It has extremely high abundance, rapidly internalizes together with any bound molecules, and recycles back to the cell surface within 15 min [30,91]. Strong binding and internalization are afforded by triantennary GalNAc ligands with optimal spatial arrangement for concurrent binding to three sites of the hetero-oligomeric receptor [92]. Consequently, sufficient oligonucleotide cargo is internalized, and high endosomal loading appears to overcome the rather inefficient endosomal escape of bioconjugates [29]. As no other receptors with such high abundance and rapid uptake and recycling kinetics have so far been identified [23], it seems necessary to improve endosomal escape mechanisms, for application of a receptor-mediated uptake strategy for targets aside of hepatocytes.
Unless better suited target receptors with high abundance and quick internalization rates are identified, rational improvement of oligonucleotide delivery arguably needs to focus on better understanding and finally improving endosomal escape. Small molecule agents [93,94], basic peptides [95], and polymers [96] for enhancing endosomal escape have been reported, for all of which potential toxicity and therapeutic index needs to be taken into account. More sophisticated strategies are pore-forming or membrane-destabilizing compounds [97]. The clinical stop of a melittin-based strategy for enhancing siRNA uptake and endosomal escape because of toxicology concerns illustrates the narrow path for any such approach [29]. Of note, the effects of most compounds reported to improve functional uptake are dependent on the precise delivery system (lipid particles, chemical modification, etc.) and cell model [94], and a safe therapeutic use of such an approach is still uncertain. In particular, compounds which rely on cationic charges have so far ultimately been hampered by in vivo toxicity.
Further Development and Optimization
For realizing the full potential of oligonucleotide therapeutics, effective solutions for extrahepatic delivery are needed. Current clinical and preclinical data strongly indicate that this can be achieved only through active targeting. Both particle-and conjugate-based approaches have their virtue, although bioconjugates seem to be the slightly superior and more versatile solution as the influence of passive targeting based on particle-size and off-target binding caused by an avidity effect will be difficult to overcome. On the other hand, saturation of receptor-mediated uptake, repulsion through negative charge, and particularly insufficient endosomal escape are bottlenecks for bioconjugates.
For solving these problems, either a drastic increase of the number of oligonucleotides internalized by each uptake event or the development of more effective and preferably specific endosomal escape strategies is necessary. Optimization of existing endosomolytic compounds may be a solution. Due to the risk of toxicity, such functionalities would preferably be combined into a single bioconjugate molecule to specifically destabilize only oligonucleotide-containing endosomes for limiting adverse effects on nontargeted cells. Nevertheless, a low therapeutic index of endosomolytic substances is likely inherent, and more sophisticated strategies might be needed.
The mechanism of protein toxins is a biological model for biomacromolecules entering the cytosol through endosomes and can be combined with receptor-mediated uptake by DARPins or antibody domains [98–100]. The toxins of cholera [101], Pseudomonas aeruginosa [102], and shiga [103], as well as ricin, are all delivered with high efficiency to the cytosol through endosomes by profiting from sorting to the Golgi network and retrograde transport to the endoplasmic reticulum (ER). Exploitation of respective mechanisms may be a viable strategy for cytosolic oligonucleotide delivery. This is however not easily implemented, as a protein cargo has completely different structural and biophysical properties than nucleic acids.
The P. aeruginosa exotoxin A (ETA) is transported from endosomes to the ER by a KDEL ER retrieval motif. However, attachment of this short peptide sequence to an oligonucleotide cargo did not meaningfully enhance endosomal escape of antisense or siRNA ([104] and unpublished data). The attachment of the oligonucleotide cargo with its multiple anionic charges may prevent the interaction of short signal peptides with their endosomal receptors by being repelled by anionic membranes. A solution may be better compound design, such as a larger protein component specifically designed for intracellular trafficking. However, when using larger peptides or proteins with oligonucleotide cargoes attached, the issue of cleavage of the carrier after successful cytosolic delivery remains as the oligonucleotide must finally be available for RISC binding or mRNA hybridization. Enzymatically cleavable or hydrolyzable linkers between individual conjugate component that are used for receptor targeting, cytosolic delivery/intracellular trafficking, and finally gene silencing could be developed, which on the other hand arguably complicates compound synthesis and stability [105].
Another viable strategy might be multimerization of nucleic acid cargoes in relation to the targeting protein. Receptor-mediated uptake is arguably quickly saturated depending on receptor kinetics, and for most bioconjugates, only a single oligonucleotide molecule enters the cell through the endosomal pathway per uptake event. With the exemption of the asialoglycoprotein receptor-dependent uptake of GalNAc conjugates, this limitation together with high endosomal retention of the cargo is insufficient for practically all cell surface receptors. Covalent linkage of more than two or three oligonucleotides to a single protein is a complicated task [106].
Nucleic acids themselves could be used as a scaffold for self-assembly of multiple effector siRNAs or antisense compounds, taking advantage of their sequence-specific hybridization, generating well-defined three-dimensional structures [107,108]. There is some evidence that tightly assembled nucleic acids even enhance passive cellular uptake and endosomal escape [109]. In contrast, when assembling more than a handful oligonucleotide, possible side effects based on sequence-specific off-target binding and immune recognition by Toll-like receptors can be triggered [110–112], and molecule size increases, in turn influencing biodistribution. Stability and stable association of hybridization-based approaches need to be ensured, possibly enhanced through chemical modification of the scaffolding nucleic acids.
In conclusion, important progress in delivery of therapeutic oligonucleotides through active targeting beyond the liver has been made. For high-affinity receptor-protein interactions, bioconjugates altogether appear to be more promising than nanoparticle approaches. While good target and tissue specificity can be already achieved, the last and most persistent hurdle of more efficient endosomal escape remains to be mastered.
Footnotes
Author Disclosure Statement
No competing financial interests exist.