
Abstract
Genetic drugs based on RNA or DNA have remarkable therapeutic potential as virtually any disease can be treated by silencing a pathological gene, expressing a beneficial protein, or by editing defective genes. However, therapies based on nucleic acid polymers require sophisticated delivery systems to deliver these macromolecules to the interior of target cells. In this study, we review progress in developing nonviral lipid nanoparticle (LNP) delivery systems that have attractive properties, including ease of manufacture, reduced immune responses, multidosing capabilities, larger payloads, and flexibility of design. LNP systems represent the most advanced delivery systems for genetic drugs as it is expected that an LNP-short interfering RNA (siRNA) formulation will receive clinical approval from the Food and Drug Administration (FDA) in 2018 for treatment of the hereditary condition transthyretin-mediated amyloidosis, a fatal condition for which there is currently no treatment. This achievement is largely due to the development of optimized ionizable cationic lipids, arguably the most important factor in the clinical success of LNP-siRNA. In addition, we highlight potential LNP applications, including targeting tissues beyond the liver and therapeutic approaches based on messenger RNA or Clustered Regularly Interspaced Short Palindromic Repeats/Cas.
Introduction
G
Alnylam® Pharmaceuticals' formulation, known as Patisiran, met all primary and secondary endpoints, and showed robust and sustained knockdown of TTR [9]. Submission of a New Drug Application to the Food and Drug Administration (FDA) was recently completed [10] and the expected approval of Patisiran in 2018 will mark the first successful systemic RNAi treatment for patients. The European Medicines Agency (EMA) recently accepted the marketing authorization application for Patisiran [11]. In addition, there are a number of LNP formulations of siRNA and other genetic drugs currently undergoing clinical evaluation for treatment of various diseases, including cancer and viral infections (Table 1).
ACC, adrenocortical carcinoma; AML, acute myeloid leukemia; CEPBA, CCAAT enhancer binding protein alpha; EphA2, ephrin type-A receptor 2; DOPC, dioleoyl-phosphatidylcholine; GI-NET, gastrointestinal neuroendocrine tumors; Grb2, growth factor receptor-bound protein 2; H7, hemagglutinin 7; H10, hemagglutinin 10; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HSP47, heat shock protein 47; KSP, kinesin spindle protein; MAAs, melanoma-associated antigens; mRNA, messenger RNA; PCSK9, proprotein convertase subtilisin/kexin type 9; PKN3, protein kinase N3; PLK1, polo-like kinase 1; siRNA, short interfering RNA; TAAs, tumor-associated antigens; TTR, transthyretin; VEGF-A, vascular endothelial growth factor A.
Lipid-based drug delivery systems represent a technology with a large knowledge base and well-understood design principles. There are now 10 LNP drugs that have received regulatory approval for delivery of small molecule drugs [12]. In this study, we review the challenges involved in extending LNP technology to nucleic acid polymers, the critical factors that have enabled progress to this point, and the remarkable way in which LNP technology developed for siRNA can also be applied to much larger payloads such as mRNA and pDNA.
Barriers to Gene Therapies Based on Nucleic Acid Polymers
Initial research in the 1990s focused on the delivery of DNA, with the aim to induce expression of a gene of interest in target cells. The half-life of functional pDNA in mouse whole blood (in vitro) was shown to be ∼10 min, and even shorter in vivo upon intravenous (i.v.) administration [13] where rapid and extensive hepatic accumulation in nonparenchymal cells were observed. This elimination from circulation was largely a result of serum nuclease activity and represents the first major barrier to delivery of nucleic acid therapeutics. The activation of innate immunity represents a second impediment [14]. In the case of mRNA and siRNA, this immune response can be partially circumvented through chemical modification of the nucleic acids [15]. On the other hand, pDNA, which is purified from biological sources rather than synthesized, is less amenable to such modifications. It also suffers from an increased likelihood of pyrogens (endotoxins) being present in the raw material. However, even if nucleic acids are heavily modified to overcome serum nuclease activity and immune stimulation, they are still subject to a third barrier, renal filtration (efficient filtration of particles smaller than ∼6 nm) [16,17]. Studies into the pharmacokinetics of oligonucleotides showed more than 50% of the dose excreted in urine [18,19]. These observations suggest that the utility of unmodified nucleic acid polymers as therapeutics is limited due to their poor bioavailability. Modifications to improve the activity of nucleic acids without delivery vectors have been reviewed elsewhere [20].
Attempts have been made to improve the bioavailability of nucleic acid drugs by predosing with negatively charged polymers such as dextran sulfate [13]. While this improves the pharmacokinetics, the nucleic acids still do not preferentially accumulate at target sites. Even if accumulation is achieved, the physicochemical properties of nucleic acids (bulky size and high charge density) represent a fourth barrier as they prevent diffusion across the cell plasma membrane. Internalization through endocytosis presents problems as the endocytic pathway ends in fusion with the lysosome, where the nucleic acid polymer would be degraded, requiring escape from the endosome to the cell cytoplasm for the DNA or RNA vector to be active. In the case of pDNA and CRISPR therapeutics, there is the subsequent barrier of entering the nucleus for activity [21] as such transport does not occur through passive diffusion unless cell division occurs where the nuclear membrane is temporarily compromised [22].
To achieve delivery across these barriers, many types of systems have been explored [2–4]. In this study, we focus on lipid-based delivery systems, and the first demonstration of lipid-based transfection was an in vitro study by Felgner et al. using the positively charged lipid, N-[1-(2,3-dioleyloxy) propyl]-N,N,N-trimethylammonium chloride (DOTMA) [23]. Compared to techniques such as calcium phosphate or DEAE-dextran-mediated transfection, “lipofection” was found to be between 5- to 100-fold more effective. This observation started the development of lipid-based vectors for nucleic acid delivery.
Design of LNPs for Delivery of Nucleic Acids
As noted, LNP formulations of small molecule drugs represent a relatively mature technology. Design features (reviewed elsewhere [12,24]) such as optimal particle sizes (100 nm diameter or less), near 100% encapsulation efficiencies, robust manufacturing processes, and low surface charge to reduce interactions with serum proteins are also required features for lipid-based delivery systems for nucleic acids. However, different loading strategies and manufacturing techniques are required. These methods must enable efficient and scalable entrapment (>80%) of nucleic acid polymer as well as an ability of the resulting LNP to deliver cargo to the cytoplasm of target cells. To achieve encapsulation of negatively charged polymers, cationic lipids are required; these lipids have gone through considerable evolution as detailed below.
Cationic lipids and lipoplexes
Early efforts to develop LNP systems relied on entrapment through passive methods using neutral (zwitterionic) lipids, which did use electrostatic properties to achieve entrapment. Such formulations displayed poor entrapment efficiencies (<40%) [25] and limited transfection potency [26]. The development of DOTMA [23] enabled more efficient entrapment and transfection potency through formation of complexes where DOTMA was mixed with a “helper lipid,” dioleoylphosphatidylethanolamine (DOPE), and complexed by mixing with DNA in solution. These complexes, termed “lipoplexes,” are unstable and characterized by broad size distributions ranging from the submicron scale to a few microns. Lipoplexes, such as the Lipofectamine® reagent, have found considerable utility for in vitro transfection. However, these first-generation lipoplexes have not proven useful in vivo. The large particle size and positive charge (imparted by the cationic lipid) result in rapid plasma clearance, hemolytic and other toxicities, as well as immune activation [27,28].
Cationic lipids and LNPs
The first example of an LNP formulation of a genetic drug (pDNA) containing a cationic lipid employed a detergent dialysis technique. The approach comprised mixing of pDNA with cationic lipids (dioctadecyl-dimethyl-ammonium chloride or DODAC) in the presence of a detergent such as octylgluopyranoside (OGP), followed by addition to a solution containing phosphatidylcholine (PC)-lipids and polyethylene glycol (PEG)-lipid in an OGP solution [29]. The final mixture was then dialyzed for 36–48 h to remove detergent and form nanoparticles. At cationic lipid contents of 6–8 mol%, entrapment efficiencies of ∼30% were achieved, but replacement of the PC-lipid with DOPE resulted in entrapment efficiencies of 70%. These particles were termed stabilized-plasmid-lipid-particles (SPLP) and displayed unilamellar structure under cryo-TEM with relatively monodisperse size distributions around 70 nm [30,31].
SPLP exhibited desired long circulation properties with t1/2 of ∼7 h in mice compared to <10 min for naked pDNA. This enabled 2.5% of the injected dose to accumulate at a distal tumor over 24 h [31], resulting in expression of a luciferase reporter in tumor tissue, with very low levels in the liver and spleen [32]. Importantly, the liver toxicity induced by SPLP was significantly lower when compared to lipoplexes generated using the same lipid mixture and lipid-pDNA ratio [31]. At a dose of 30 μg DNA, lipoplexes induced a 100-fold increase in levels of liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in serum, while SPLP at a dose of 175 μg DNA showed baseline levels of ALT and AST in serum. However, given the low encapsulation efficiency, limited potency, and the fact that the detergent dialysis manufacturing technique is tedious and not scalable, the SPLP approach was not pursued further.
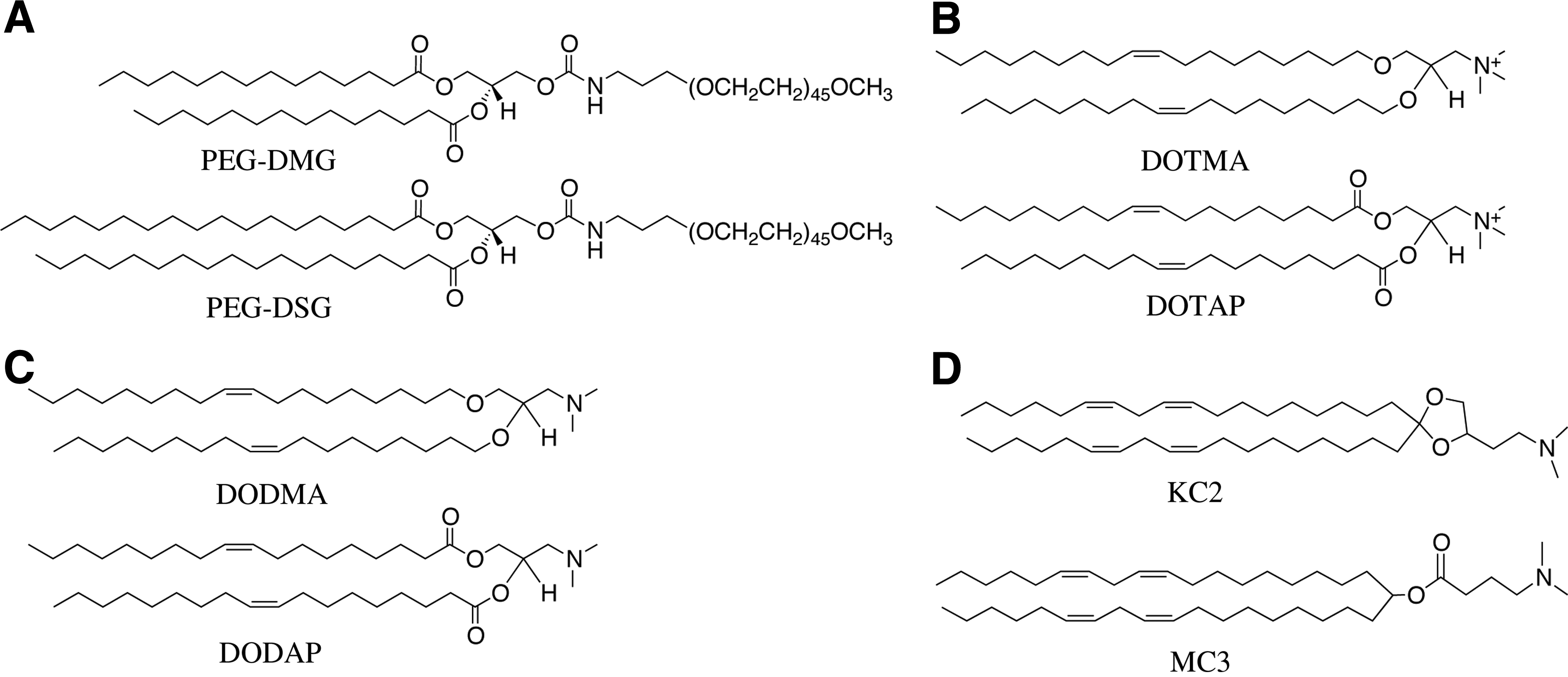
Efficient encapsulation of high levels of nucleic acid polymers in LNP systems intuitively requires high levels of cationic lipids to associate with the negatively charged polymers. However, cationic lipids have inherent limitations for in vivo use. LNPs with a significant surface charge adsorb serum proteins and are rapidly cleared from the circulation [12]. Furthermore, cationic lipids combine with endogenous anionic lipids to form nonlamellar HII phase structures [33], thus disrupting cell membranes and accounting partially for their toxicity. Another contribution to toxicity results from the production of reactive oxygen species [34–36]. Cationic lipids, however, are required to achieve efficient loading of nucleic acid polymers into LNPs. To address this problem, ionizable cationic lipids such as 1,2-dioleoyl-3-dimethylammonium propane (DODAP) were developed with apparent pKa values below 7. LNPs could then be loaded with nucleic acid polymers at pH values below the pKa of the ionizable lipid where it is positively charged. However, at physiological pH values, the LNP would adopt a relatively neutral exterior [37]. Development of lipids such as 1,2-dioleyloxy-N,N-dimethyl-3-aminopropane (DODMA) or DODAP (Fig. 1) provided the near-neutral charge in circulation, allowing for significant increases in the circulation half-lives of the particles following i.v. administration [38].
The evolution of lipids to enable gene silencing in vivo.
Ionizable Cationic Lipids and Ethanol Loading
The detergent-dialysis loading process, while effective for producing homogenous LNP, is laborious. Thus, ethanol loading processes were devised for use in combination with ionizable cationic lipids. The first version involved dissolving lipids in ethanol and adding this solution to an aqueous solution of antisense oligonucleotides (ASO) in pH 4 buffer [38]. The resulting particles were then extruded to achieve a homogenous population of ∼100 nm LNP, followed by dialysis to remove solvent and neutralize the pH. This process resulted in entrapment efficiencies of 80% for a lipid composition of DODAP/DSPC/Cholesterol/PEG-ceramide-C14 (20/25/45/10 mol%). In the next iteration [39], liposomes were initially formed by extrusion and then subjected to ethanol concentrations as high as 40% (v/v) in the presence of ASO at acidic pH. The resulting particles displayed entrapment efficiencies as high as 90% for the ASO and 70% for pDNA systems. The inclusion of at least 2.5 mol% PEG-lipid resulted in stable particles, although up to 10 mol% PEG-lipid was required to maintain particle size through the incubation.
The final iteration of the ethanol loading process involved the use of a spontaneous vesicle formation process [40] following the Batzri and Korn ethanol injection method [41]. In this process, the desired lipids are dissolved in ethanol at appropriate ratios, while pDNA is dissolved in an acidic buffer. The solutions are then rapidly mixed through a T-junction [42] with an output of 1 mL/s where the ethanolic phase contains lipid at a concentration of ∼20 mM. The resulting nanoparticles were then immediately put through another T-junction to dilute the ethanol concentration. After the first mixing step, the entrapment was determined to be ∼60%, increasing to 80%–90% after the second dilution. An improvement to this loading procedure relied on the combination of the two mixing steps into a single step to achieve a final concentration of 25% ethanol (v/v), although the mixing was performed with a microfluidic mixing device with an output of 2 mL/min, resulting in an effective 30-fold reduction in output [43]. This method of diluting lipid-in-ethanol into an aqueous buffer by rapid mixing is now the formulation technique of choice. These methods have been reviewed in detail elsewhere [44]. The method achieves effectively complete encapsulation and has the added advantage of being relatively gentle as required by nucleic acid polymers. Techniques requiring high temperature and high shear forces likely cannot be used.
Ionizable Cationic LNPs for siRNA Delivery
Application of the ethanol loading technique in combination with ionizable cationic lipids such as DODAP resulted in LNP formulations of ASO that had potential in vivo applications. However, efforts to use LNP systems to improve ASO gene silencing potency had limited success, while immunostimulatory applications had more potential, although this has not yet been exploited [45–47]. The most compelling application, however, was for siRNA. Using the dilinoleoyl version of the ether analogue of DODMA, Dilinoleyl-DMA (DLinDMA), it was found that LNP-siRNA systems resulted in significant gene silencing in the liver following i.v. administration [48]. These authors used the ethanol loading T-junction process and the lipid composition DLinDMA/cholesterol/DSPC/PEG-lipid (30/48/20/2 mol%) to formulate LNP ApoB-siRNA and showed dose-dependent silencing of ApoB in the liver 48 h after a single injection, with maximal silencing of >90% in nonhuman primates (cynomolgus monkeys) at doses of 1 or 2.5 mg/kg. Significant reductions in ApoB protein, serum cholesterol, and low-density lipoprotein levels were observed as early as 24 h after treatment and lasted for nearly 2 weeks at the highest dose.
The type of PEG-lipid used in the ApoB silencing study was termed “diffusible” PEG-lipids, as in the presence of a lipid sink (such as serum lipoproteins), the PEG-lipids were able to diffuse out of the nanoparticle in times on the order of minutes and expose the surface of the LNP [32]. Diffusible PEG-lipids had shorter acyl chains (C14; (R)-2,3-bis(myristoyloxy)propyl-1-(methoxy poly(ethylene glycol)2000) carbamate, abbreviated as PEG-DMG) than the persistent PEG-lipids (C18; (R)-2,3-bis(stearyloxy)propyl-1-(methoxy poly(ethylene glycol)2000 carbamate, abbreviated as PEG-DSG) that have extended residence times used for LNP systems where long circulation lifetimes are required (Fig. 1). The use of the diffusible PEG-lipids to dissociate the PEG coating renders the LNP transfection competent and contributed significantly to improving transfection. The evolution of these PEG-lipids has been reviewed elsewhere [49], but it is important to note that the PEG-lipid is essential to maintaining the size and stability of the LNP before administration. Furthermore, the use of diffusible PEG lipids mitigates against an immune response to LNP-associated PEG as they do not remain associated with the LNP [50].
Design of ionizable cationic lipids and clinical applications
The initial reason for employing ionizable cationic lipids was to achieve a system where efficient encapsulation of negatively charged polymers could be achieved at low pH, but which also exhibited a relatively uncharged surface at pH 7.4. However, it soon became clear that the potency of LNP-siRNA systems for silencing genes in hepatocytes following i.v. administration was sensitive to the species of ionizable cationic lipid employed. Thus, a comprehensive screening process was undertaken to see whether further improvements over the gene silencing achieved by LNP-siRNA systems in the liver (hepatocytes) were possible. A guiding hypothesis was that the most potent ionizable cationic lipids would satisfy two constraints. The first constraint is that the pKa of the lipid should be low enough to prevent the LNP from having a high positive surface charge at physiological pH values. High surface charge is associated with toxicity and rapid clearance from the circulation by the fixed and free macrophages. The second constraint is that the pKa should be high enough that the ionizable cationic lipid can adopt a positively charged form at acidic endosomal pH values. This is so that cationic lipids can combine with endogenous endosomal anionic lipids to promote membrane lytic nonbilayer structures such as the hexagonal HII phase, resulting in intracellular delivery (Fig. 2). It is well known that cationic lipids combine with anionic lipids to adopt nonbilayer structures and lipids that adopt such structures have significant membrane-destabilizing properties [33,51,52]. A related design principle was that the cationic lipid should be relatively unsaturated as it is well established that unsaturated lipids adopt these nonbilayer structures more readily [53,54].

Mechanism of transfection with an LNP formulation of siRNA. LNP transfection following endocytosis is mediated by an endosomolytic event where the contents of the nanoparticle are delivered to the cytosol of the target cell.
The polymorphic phase tendencies of lipids can be rationalized according to the “shape” hypothesis [54] where a comparison of the cross-sectional area of the lipid head group to the cross-sectional area of the lipid tails determines the macrostructure formed upon hydration (Fig. 3). Lipids such as lysolipids and PEG-conjugated lipids with a “cone-shaped” geometry prefer to form micellar structures. Alternatively, lipids such as distearylphosphatidylcholine (DSPC) have a cylindrical geometry compatible with bilayer structures. Other lipids with small uncharged head groups and unsaturated acyl chains, such as DOPE, display an “inverted-cone” geometry, and preferentially form the inverted hexagonal (HII) phase [53].
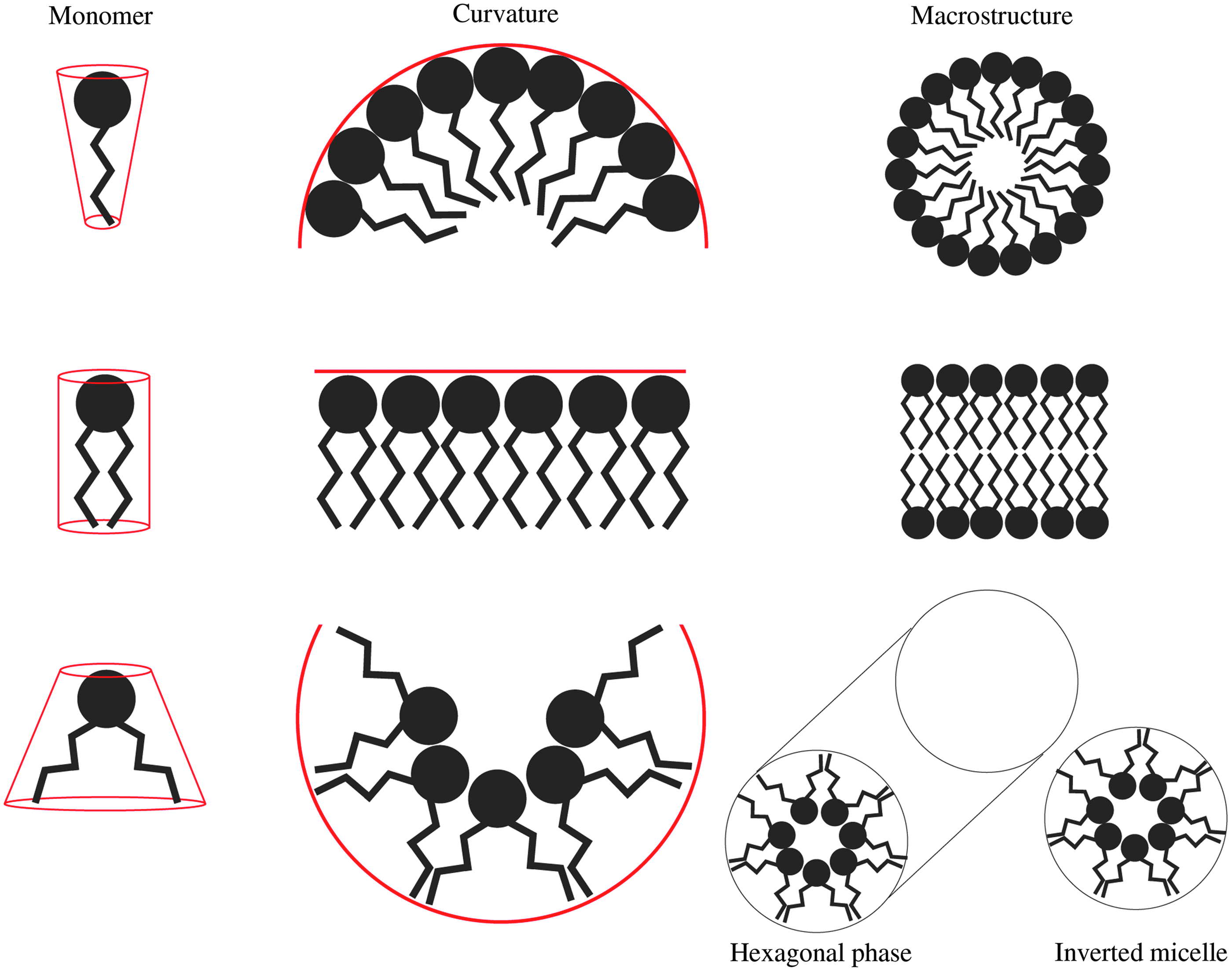
The molecular shape hypothesis and lipid polymorphic behavior. The molecular shape hypothesis suggests that the macrostructure adopted by a lipid is dictated by the geometry of the lipid. This geometry is defined by the cross-sectional area of the lipid head group and cross-sectional area of the lipid tails. (Top) A cone-shaped lipid (large head group, small tail area) generates positive curvature in membrane, and in isolation will form micelles. (Middle) A cylindrical-shaped lipid does not generate curvature, and in isolation will form bilayer structures. (Bottom) A lipid with an inverted cone geometry induces negative curvature and upon hydration forms the hexagonal (HII) phase, inverted micelles, and cubic phases (not shown).
Using a murine Factor VII (FVII) model for hepatic gene silencing, LNPs containing a variety of ionizable cationic lipids were evaluated to determine the effective dose to achieve 50% gene silencing (ED50). The rational-design process first identified 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA, or KC2) as an ionizable cationic lipid that had improved activity compared to DLinDMA [8]. Further optimization of the LNP-siRNA systems was achieved by increasing the lipid content to 50 mol% [8], resulting in an ED50 for KC2 of 0.1 mg siRNA/kg with no apparent hepatic toxicities. A subsequent screening study identified heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino)butanoate (DLin-MC3-DMA or MC3) [7]. The structures of the ionizable cationic lipids discussed are shown in Fig. 1. The optimized lipid formulation of MC3/cholesterol/DSPC/PEG-lipid (50/38.5/10/1.5 mol%) in the same FVII assay displayed an ED50 of 0.005 mg/kg.
MC3 represents the most potent ionizable cationic lipid thus far identified for hepatic gene silencing. It was determined that lipids such as MC3 and KC2 exhibited optimal apparent pKa values in the region of 6.5. This pKa activity correlation was confirmed by mixing ionizable lipids with different pKa values to achieve net pKa values ranging from 5.6 to 6.9. LNP-siRNA containing lipids with a net pKa of ∼6.5 displayed optimal silencing in the murine FVII model [7]. Overall, it should be noted that optimization of ionizable cationic lipids led to a truly remarkable improvement in gene silencing potency. Initial LNP-siRNA systems composed of DLinDAP exhibited an ED50 of 40–50 mg/kg, while the ED50 for MC3-LNP was 0.005 mg/kg, an improvement of more than three orders of magnitude with no increase in toxicity. This corresponds to a therapeutic index improvement greater than 8,000-fold, suggesting clinical utility.
As a result, LNP-siRNA systems containing MC3 [7,55,56] rapidly transitioned into clinical trials conducted by Alnylam Pharmaceuticals to treat transthyretin (TTR)-induced amyloidosis, a hereditary disease resulting from mutations in the TTR gene, leading to deposition of TTR fibrils in cardiac and nerve tissue. TTR is synthesized predominantly in hepatocytes and thus susceptible to the gene silencing possible with LNP-siRNA systems containing MC3. A Phase I clinical evaluation of the formulation indicated that the LNP-siRNA was well tolerated at doses up to 0.5 mg siRNA/kg [57]. A subsequent phase II study determined that only 0.3 mg siRNA/kg body weight in a dosing regimen of Q3 W was required to achieve robust gene silencing, with an excellent tolerability profile [58]. It should be noted that as lipid-based drug delivery systems have been associated with adverse infusion-related immune events [59], a mixture of immune suppressants is given before LNP-siRNA [58]. Patisiran recently (September 2017) completed a Phase III clinical trial that met all primary and secondary endpoints [9] and exhibited less toxicity than the control substance (sterilized normal saline). A New Drug Application for regulatory approval of Patisiran was made to the FDA in December 2017.
Structure of LNP-siRNA formulations
LNP-siRNA systems containing KC2 and MC3 exhibited an electron-dense core structure as visualized by cryo-TEM [60] compared to the clearly lamellar structure of certain LNP systems containing plasmid [31] or ASO [39]. This electron-dense core structure has been suggested to result from the initial formation of inverted micellar structures of cationic lipids surrounding siRNA molecules that are subsequently coated with polar lipids, such as the PEG and phospholipids, as the polarity of the medium increases [43,60] (Fig. 4). This proposed LNP structure has been supported by a number of findings such as the dependence of particle size on PEG-lipid content, 31P-NMR studies showing immobilization of the nucleic acid, the “currant bun” morphology of entrapped negatively charged gold nanoparticles, and computer simulations. These findings and their contribution to the suggested structure are briefly discussed in turn:

Proposed mechanism of LNP formation and nanostructured core. Increase in solvent polarity (light gray line, background) drives the self-assembly of LNP-siRNA formulations. LNP-siRNA are hypothesized to form an electron-dense core structure as a result of significant lipid and nucleic acid present in the internal compartment. (Left) The first interactions to occur upon mixing of the ethanol and aqueous streams are those between the ionizable cationic lipids and the negatively charged nucleic acids. (Middle left) As the solvent polarity progressively increases, the hydrophobic inverted micellar structures coalesce generating the core of the LNP. (Middle right) As mixing continues, the more polar lipids (such as PEG-lipid and DSPC) coat the surface of the nanoprecipitates. (Right) The resulting part has an electron-dense core structure surrounded by a lipid monolayer. Ionizable cationic lipid (purple), phospholipid (dark gray), PEG-lipid (violet), cholesterol (light gray), and siRNA (red). DSPC, distearylphosphatidylcholine.
(1) It was determined by varying the concentration of PEG-lipid (at the expense of cholesterol) that particle diameters could be varied from 120 nm (at 0.25 mol% PEG-lipid) to 25 nm (at 5 mol% PEG-lipid) [43]. These observations were consistent with the PEG-lipid being present in the surface monolayer and thus dictating LNP size by influencing the surface-to-core lipid ratio.
(2) “Limit size” refers to the smallest structures compatible with a particular lipid composition. The formation of limit-size particles was achieved through formulation of KC2/PEG-lipid (90/10 mol%) with siRNA generating 23 nm particles [60]. This size corresponds to the theoretical size of a single inverted micellar structure coated with a surface monolayer of lipids [60];
(3) 31P-NMR data of phosphorothioate-modified siRNA (to prevent overlap from DSPC-phosphate signal) showed a loss of signal when siRNA was entrapped in LNP systems, showing that the inverted micellar structures immobilize the siRNA on the NMR timescale [60];
(4) The entrapment of gold nanoparticles through a different charge density provided a “tracker” for analysis by cryo-TEM due to the electron density differences. These formulations displayed a “currant bun” morphology, which strongly supports the inverted micellar hypothesis [61];
(5) Computer simulations of siRNA interactions with ionizable cationic lipids suggested periodic compartments in which the siRNA could be immobilized [60];
LNP-siRNA protein adsorption and endogenous targeting to hepatocytes
The relative specificity of LNP-siRNA systems containing ionizable cationic lipid for hepatocytes suggested that a targeting mechanism is operating. Given previous results [62] demonstrating that clearance of neutral liposomes to hepatocytes is mediated by ApoE, its role in delivery of LNP-siRNA systems was investigated. These studies demonstrated that LNP-siRNA gene silencing activity was significantly decreased in an ApoE knockout mouse model (ApoE−/−) and activity could be rescued by preincubating the particles with ApoE before administration [63]. It was therefore concluded that ApoE association with LNP-siRNA systems plays a major role in triggering LNP uptake into hepatocytes. It appears that the LDL receptor plays a role in the ApoE-dependent uptake. In a low-density lipoprotein receptor knockout model (LDLR−/−), LNP-siRNA formulations displayed less gene silencing activity (leading to higher ED50 values) than in wild-type animals [63]. LNP activity could be rescued through addition of a multivalent targeting ligand, N-acetylgalactosamine (GalNAc), for the hepatocyte asialoglycoprotein receptor, thereby promoting internalization through an alternative endocytic route.
ApoE-binding to LNP-siRNA systems subsequent to the dissociation of the PEG-lipid coating clearly plays a role in the rapid particle accumulation in the liver following i.v. administration [64]. A study with a variety of LNP-siRNA formulations showed that 95% of the injected dose of LNP with the composition ionizable lipid/cholesterol/DSPC/PEG-lipid (50/38.5/10/1.5 mol%) accumulated in the liver within 15 min [64]. The size of the LNP system also plays a role in determining potency with LNP with diameter ∼80 nm exhibiting maximum gene silencing potency [64]. Smaller sizes (∼25 nm diameter) achieved by increasing the PEG-lipid content to 5 mol% resulted in significantly reduced gene silencing activity using the FVII model. The ED50 increased from 0.05 mg siRNA/kg for the 80 nm diameter to 0.8 mg siRNA/kg for the 25 nm diameter LNP-siRNA system. This reduced activity was attributed [64] to instability of the smaller particles leading to dissociation of the cationic lipid from the particle. The reduced activity could be partially recovered by increasing the ionizable cationic lipid content to 60 mol% and increasing the ionizable lipid to siRNA charge ratio (N/P) from 3 to 6, resulting in an improvement of the ED50 from 0.8 mg siRNA/kg to 0.3 mg/kg [64].
Future Perspectives
Clinical validation of LNP-siRNA systems to silence genes such as TTR in hepatocytes clearly opens the door to using similar systems to silence other genes in hepatocytes such as proprotein convertase subtilisin/kexin type 9 (PCSK9) for cholesterol-lowering therapy [65]. Important additional opportunities include the use of LNP systems to silence genes in tissues other than the liver and the extension of LNP technology to delivering mRNA and CRSPR-Cas for gene expression and gene editing purposes. The ability to extend gene silencing to nonhepatic tissues will dramatically extend the range of gene therapies possible, including treating diseases such as cancer by silencing oncogenes. LNP-mRNA systems, on the other hand, offer considerable potential for expressing therapeutic proteins to treat a wide variety of diseases requiring protein replacement as well as vaccine applications. Finally, the ability to practice gene editing in vivo would clearly enable treatment of many hereditary diseases. A number of these opportunities are now being evaluated in clinical settings (Table 1).
Design of LNP-siRNA for extrahepatic targets
Liposomal systems used for the delivery of small-molecule therapeutics often employ a persistent PEG-lipid coat to increase circulation times, resulting in improved delivery to nonhepatic tissues such as tumor sites [12]. However, incorporation of persistent PEG-lipids such as PEG-DSG achieved the desired circulation times for LNP-siRNA systems [64], but dramatically reduced transfection competency, and as observed with a number of cationic lipid systems, can completely abolish it [66,67]. Thus, while extrahepatic distributions of LNP containing persistent PEG-lipids can be achieved, the LNP reaching these tissues have limited transfection potency. This “PEG-dilemma” has yet to be solved.
The situation is further complicated by the dual role of the PEG-lipid in LNP-siRNA formulations, serving to control particle size [43] as well as circulation lifetimes in vivo [64]. LNP-siRNA systems containing 1.5% PEG-DSG exhibit a size of 45 nm, but display rapid clearance (t1/2 < 15 min) and reasonable gene silencing potency, presumably because they retain the ability to associate with ApoE at these relatively low PEG contents [64]. In contrast, LNPs containing 5 mol% PEG-DSG and a size of 25 nm exhibit t1/2 of ∼10–12 h [64]. The LNP size can also influence circulation time in another way; particles with 0.5 mol% PEG-DSG and a size of 80 nm have a longer circulation t1/2 of 3–4 h [64], presumably because these larger systems are less able to pass through the fenestration of liver vasculature, leading to longer circulation lifetimes [64]. Higher (5 mol%) PEG-DSG levels leading to small particles of 25 nm stay in circulation for longer as they have a high PEG density precluding adsorption of serum proteins such as ApoE [64].
LNP-siRNA systems containing PEG-DSG (3–5 mol%; 30 nm diameter) that exhibit long circulation lifetimes (t1/2 of 10–12 h) have been used to knock down the androgen receptor (AR), a fundamental driver of prostate cancer, in an LNCaP xenograft model [68]. However, the dose of LNP-siRNA required to achieve appreciable gene silencing was 30 mg/kg, more than 6,000 times higher than required to achieve an ED50 in hepatocytes. In an effort to improve potency, a targeting ligand for the prostate-specific membrane antigen was incorporated, resulting in improved gene silencing activity [68], but not the orders of magnitude improvement required for clinical potential. Similarly, knockdown activity and tumor growth delay were reported at distal tumors in an enzalutamide-resistant LNCaP xenograft model when LNP-siRNA against clusterin was used in combination with AR-ASO [69]. The aggressive dosing regimen in these studies resulted in a cumulative dose of 30 and 50 mg siRNA/kg, respectively. While these doses were tolerated in preclinical studies, the dose levels employed were close to maximum tolerated doses, leading to a small therapeutic index that does not warrant clinical development.
In the central nervous system, ApoE is mainly produced by astrocytes [70] and transports cholesterol to neurons through ApoE receptors, which are members of the low-density lipoprotein receptor gene family. Thus, LNP-siRNA systems identical to those employed for silencing genes in hepatocytes can be used to silence genes in neurons. However, nanoparticles such as LNP-siRNA do not passively cross the blood–brain barrier, requiring local administration. The first demonstration of LNP-siRNA activity in neurons was targeting the GluN1 subunit of the N-methyl-D-Aspartate receptor following intracortical and intracerebroventricular administration [71]. Intracortical administration resulted in silencing around the injection site, while intracerebroventricular administration resulted in more widespread knockdown. Subsequent studies with LNP-siRNA enabled the elucidation of a secondary role of the ion exchanger SLC26A11 as a voltage-gated ion channel involved in neuronal swelling and cytotoxic edema [72]. Suppression of SLC26A11 by LNP-siRNA-mediated knockdown dramatically decreased neuronal swelling. These studies highlight the potential of LNP-siRNA for target identification studies, as well as the potential for therapeutic applications for severe disorders where direct administration through intracortical, interventricular, or intrathecal routes is possible.
Design of LNP for gene expression and editing
The initial design of LNP systems (SPLP) for larger nucleic acid polymers was for delivery of pDNA to express a therapeutic gene. As noted previously, low expression levels and an inefficient and nonscalable formulation technique precluded further development. Following the development of the ethanol loading technique for encapsulating siRNA into LNP, it was shown that this protocol could also be used to encapsulate mRNA, pDNA, and other negatively charged macromolecules [61,73]. This has led to significant advances to enable mRNA-based therapeutics. Extensive work has been conducted to develop ionizable cationic lipids optimized for mRNA cargoes [74], leading to improvements in gene expression in hepatocytes following i.v. administration that are some 20-fold higher than observed for LNP mRNA systems containing MC3. Intravenous administration of LNPs containing unmodified, sequence-engineered mRNA encoding for erythropoietin (EPO) that was administered intravenously at a single dose of 1.3 mg mRNA in pigs (20 kg) and a dose of 100 μg mRNA in cynomolgus monkeys resulted in high systemic EPO levels, increased reticulocyte numbers, and elevation of the hematocrit [75]. This has led to the possibility of using the liver as a bioreactor to express therapeutic proteins. A compelling example is the demonstration that i.v. administration of LNP-mRNA encoding VRC01 (a broadly neutralizing antibody against HIV-1) results in high levels of the VRC01 antibody in the circulation for 7 days in humanized mice. Such levels were sufficient to afford complete protection against a challenge with the SF162 strain of HIV 24 h later. This protection required as little as 15 μg mRNA per animal [76].
LNP-mRNA systems also show considerable potential for vaccine applications. Initial work in mice showed that LNP-mRNA formulations at doses of 5 μg nucleoside-modified mRNA administered through subcutaneous, intradermal, or intramuscular injection could result in sustained expression of luciferase for up to 10 days, whereas i.v. administration resulted in a burst of expression followed by rapid decrease over 5 days [77]. A significant body of data has been published on the use of mRNA for vaccine applications [78]; certain studies employing LNP systems are of particular interest [76,79,80]. LNP-mRNA formulations encoding the premembrane envelope protein prM-E derived from the Zika virus were tested in mice and rhesus macaques and showed that a dose of only 50 μg mRNA was required to protect against a Zika virus challenge [80]. Kranz et al. have demonstrated the application of lipid-based delivery of RNA to dendritic cells for cancer immunotherapy [81]. It was shown that i.v. administration of RNA lipoplexes resulted in antigen expression in lymphoid-resident DCs. In addition, lipoplexes containing RNA encoding viral or mutant neoantigens or endogenous self-antigens induced strong antitumor responses in mouse models of melanoma, colorectal cancer, and lung cancer [81]. The safety and tolerability of the RNA lipoplexes encoding four tumor antigens (NY-ESO-1, MAGE-A3, tyrosinase, and TPTE) are currently under evaluation in a Phase I clinical trial (NCT02410733, Table 1).
With regard to LNP formulations of pDNA, the application of the ionizable cationic lipid approach is leading to potent transfection systems, but the lipid compositions for achieving maximum transfection differ substantially from those employed for siRNA. For example, KC2-based LNP demonstrated significantly better in vitro transfection than MC3-based formulations [82], and unsaturated “helper” lipids such as dioleoyl-phosphatidylcholine, stearoyl-oleoyl-phosphatidylcholine, and DOPE improved transfection potency. Optimized LNP-pDNA formulations showed robust, mosaic transfection (with no detectable toxicities) of the developing chicken limb-bud following direct in ovo injection [82].
CRISPR-based genome editing has generated much excitement due to the prospect of potentially permanently curing genetic disorders [83]. However, the safe and efficient nonviral delivery of the gene-editing components (Cas protein and a single guide RNA, sgRNA) in vivo have proven to be a considerable challenge so far [5,84]. Most recently, Yin et al. developed LNP for delivery of Cas9 mRNA and sgRNAs (LNP-CRISPR) targeting the mouse PCSK9 gene for treatment of hypercholesterolemia [85]. A single injection of LNP-CRISPR in mice resulted in undetectable PCSK9 serum levels, over 80% editing in the liver and a reduction of total cholesterol by 35%–40% [85]. Results of a 12-month study in mice with LNP-CRISPR for editing of the TTR gene were recently announced by Intellia Therapeutics. Results showed ∼97% maintained reduction in serum TTR following a single dose and around 70% editing at the target DNA site in hepatocytes [86].
Summary
The discovery of ionizable cationic lipids has proven to be a critical factor in the development of clinically viable LNP-siRNA treatments. Potent lipids such as MC3 allow efficient encapsulation of siRNA, result in a relatively neutral LNP surface charge in the circulation, and are able to promote escape of the siRNA into the cytosol following endocytosis. With the likely FDA approval of the first LNP-siRNA formulation for treatment of TTR-mediated amyloidosis in 2018, there are high clinical expectations for this new class of drugs for silencing disease-causing genes in the liver and other tissues. In addition, LNP containing ionizable cationic lipids are facilitating the rapid development of other gene therapies based on mRNA and CRISPR/Cas. Important issues remain, such as cost-effectiveness, toxicity, and off-target effects; however, the fact that LNP formulations of genetic drugs can now facilitate the knockdown, expression, or editing of virtually any gene in hepatocytes, with the promise of extension to other tissues, constitutes a major breakthrough in the development of (personalized) gene therapies.
Footnotes
Acknowledgments
The work of JAK and PRC is funded by a foundation grant (FDN 148469) from the Canadian Institutes of Health Research (CIHR). RvdM is supported by a VENI Fellowship (# 14385) from the Netherlands Organization for Scientific Research (NWO).
Author Disclosure Statement
No competing financial interests exist.