
Abstract
Keratinocyte growth factor (KGF) plays a central role in wound healing as it induces cell proliferation and motility. The use of growth factors such as KGF is therefore viewed as a promising approach in wound therapy, although effective application remains a major problem because of inactivation and the resulting short half-life of applied growth factors in wound beds. Therefore, the rational of this study was to develop and investigate an innovative strategy to improve wound healing using an in vitro-transcribed modified KGF messenger RNA (mRNA). After transfection of cells, we evaluated the effects of the produced KGF protein on cell migration and reepithelialization of keratinocytes using a scratch assay. The results demonstrate that KGF-mRNA-transfected cells exhibited a high KGF protein release that is sufficient to significantly improve reepithelialization in the performed scratch assays. Transfection with growth factor mRNA therefore seems to be a promising therapeutic strategy, especially for difficult wounds, as it leads to a temporary increase of growth factor expression in the treated wound area without interfering with the DNA of the nucleus, as seen in gene therapeutic applications.
Introduction
W
Another approach to increase the concentration of certain growth factors in wounds is gene therapy; however, this approach results in the permanent expression of a specific protein of interest after DNA incorporation into the host genome [6,10]. After cellular transfection, the increased expression of these growth factors should improve wound healing that was successfully shown in previous in vivo studies [11,12]. However, gene therapy is associated with serious safety concerns, including insertional mutagenesis and immune responses [13–16].
To circumvent these problems, messenger RNA (mRNA) therapy might provide an alternative to locally increase the concentration of specific growth factors in the wound area. Contrary to gene therapy, the effect of mRNA is only transient, which could be an important advantage in preventing malignant diseases, as previously mentioned [17,18]. Moreover, it is known that growth factors play an important role in tumorigenesis [14,15]. With mRNA therapy as an alternative, it could be possible to induce a transient yet sufficiently elevated concentration of growth factors that enhances wound healing without affecting tumorigenesis because of its short-acting effects in cells.
Therefore, the aim of our study was to investigate whether it is possible to induce the expression of a specific growth factor (ie, keratinocyte growth factor [KGF; fibroblast growth factor 7 (FGF-7)] after the transfection of cells with an in vitro-transcribed (IVT) modified KGF-encoded mRNA. KGF that is synthesized by dermal fibroblasts and other connective tissue cells under physiological conditions [19] was chosen because it seems to be an important growth factor in wound healing [19,20] and induces the proliferation and migration of keratinocytes without affecting their differentiation [21–25]. Furthermore, KGF expression is highly elevated after tissue damage (more than 150-fold induced within 24 h after injury), even in comparison with other growth factors in an in vivo mice wound healing model [26]. Our data suggest that cells transfected with IVT KGF-mRNA revealed a much higher release of KGF protein than nontransfected cells or cells transfected with a control mRNA. The amount of KGF was sufficient to promote cell migration in a simple two-dimensional scratch wound healing assay. In conclusion, mRNA therapy could be a promising strategy to improve the bioavailability of growth factors in the wound areas.
Materials and Methods
Cell culture
As human skin mainly consists of fibroblasts and keratinocytes, we used the human HaCat keratinocyte cell line (CLS Cell Lines Service GmbH) and human foreskin fibroblast (HFF) for our experiments in this study. HaCat cells have been reported to be nontumorigenic and to maintain full epidermal differentiation capacity [27]. Cells were grown in 175 cm2 cell culture flasks in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Inc.) at 37°C and 5% CO2. When the cells became confluent, the medium was removed, cells were washed twice with phosphate-buffered saline (PBS) without Ca2+ (Thermo Fisher Scientific, Inc.), and then detached using 0.25% trypsin in buffered ethylenediaminetetraacetic acid (Thermo Fisher Scientific, Inc.). Detachment was stopped with supplemented medium. After centrifugation and resuspension, cells were counted with the CASY® cell counter (Schärfe System).
mRNA synthesis
As previously described [28,29], the synthesis of modified mRNA is a multistep process, in which at first the specific protein coding sequence of a plasmid is amplified with polymerase chain reaction (PCR) and after that the DNA is transcribed into mRNA using IVT. The KGF-DNA plasmid was purchased from Eurofins Genomics. The synthesis of mRNA was carried out according to the manufacturer instructions using the HotStar HiFidelity Polymerase Kit from Qiagen and the T7 MEGAscript Kit from Ambion for IVT. The results of PCR and IVT were controlled with gel electrophoresis using a 1% agarose gel and the peqGold Range Mix DNA ladder and the 0.5–10 kb RNA ladder from Invitrogen.
Transfection of cells with KGF-mRNA
For each experiment, cells were seeded with a density of 150,000 cells/well onto a 12-well plate and incubated for 1 day. Before transfection, the KGF-mRNA (0.5 or 1 μg) was complexed with 2 μL Lipofectamine® 2000 (Invitrogen) and 500 μL OptiMEM (Invitrogen) for 20 min at room temperature. Afterward, the cells were washed once with PBS and left untreated, treated with 2 μL Lipofectamine 2000 only, or with 0.5 or 1 μg of complexed KGF-mRNA. After an incubation time of 4 h, cells were washed again with PBS and 1 mL supplemented cell culture medium was added. After 24 h, the supernatant was removed (supernatant 1) and 1 mL new supplemented medium was added. After another 24 h, the supernatant was removed again (supernatant 2), so that supernatant 1 contains all KGFs expressed within the first day after transfection and supernatant 2 contains all KGFs expressed on the second day after transfection. In addition, cells were trypsinized after removal of supernatant 2 and resolved in 1 mL medium to assess cell number and viability using the CASY Cell Counter.
KGF enzyme-linked immunosorbent assay
To determine the concentration of expressed KGF after mRNA transfection in the supernatants, we used the human FGF-7 enzyme-linked immunosorbent assay (ELISA) Kit from Sigma-Aldrich®. ELISA was performed according to the specifications of the manufacturer.
Indirect KGF-mRNA treatment of scratched cells
Scratch assay
The scratch assay is a very well-established and reproducible assay commonly used for investigating cell proliferation and migration in vitro [30]. HaCat cells were seeded with a density of 800,000 cells/well onto a 24-well plate in 1 mL medium. After 1 day of incubation, the medium was replaced by medium containing only 0.1% FBS to reduce cell growth. After another day, when cells reached complete confluency, the cell monolayer was wounded using a sterile 10–100 μL pipette tip. Afterward, cells were washed several times with PBS until all remaining cell debris was removed. Finally, 300 μL supernatant 1 or 2 from previously performed transfections was added and the scratch analyzed (t0) using a microscope (Axiovert 135 + Axiocam, objective 5×, camera 0.63× with Axiovision Release 4.8.2 software from Carl Zeiss Microscopy, Jena, Germany). After 24 and 48 h, further pictures of the same view frame were captured in duplicates. The size of the wound was determined using the TScratch software (CSElab; ETH Zürich), a computer program specifically designed for automated analysis of monolayer wound healing assays [31]. To prove that all groups have comparable baseline conditions, wound size (%) at t0 was calculated. Wound closure was calculated for each day after the start of the study [eg, wound sizet24 % − wound sizet0 % = wound closureday1 pp (percentage points)]. When the scratch assay was terminated after 48 h with HaCat supernatants or 72 h with HFF supernatants, cell viability was further investigated using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
MTT assay
The MTT assay is a colorimetric assay for assessing cell metabolic activity and viability. It is based on the quantitative detection of purple formazan that is converted by viable cells with active metabolism out of MTT [32]. Cells were washed with PBS and 150 μL DMEM (without phenol red) and 15 μL of an MTT solution (5 mg MTT/1 mL PBS) was added. After an incubation time of 4 h, all reactions were stopped by the addition of 150 μL dimethyl sulfoxide (DMSO). Finally, 2 × 100 μL of this supernatant was pipetted into a 96-well plate and absorption was measured at 540 nm.
Combined transfection and scratch assay
HaCat cells were seeded with a density of 400,000 cells/well onto a 24-well plate in 1 mL medium (DMEM + 5% FBS + 1% penicillin/streptomycin). After 1 day of incubation cells were transfected as described previously with either 0.5 or 1 μg KGF- or enhanced green fluorescent protein (eGFP)-mRNA with a medium only and a medium + Lipofectamine group as controls. After 24 h, the supernatant was removed and a scratch assay was performed like described previously. Pictures of the same view frame were captured at t0, t24, and t48 with 4× magnification and analysis was performed with TScratch as described. The supernatant was centrifuged to remove remaining cell debris and 300 μL was aliquoted for KGF ELISA, whereas the rest was pipetted onto the cells again. After 1 day, another 300 μL was aliquoted for the KGF ELISA. Twenty-seven hours after the start of the scratch assay cell viability was evaluated.
Sulforhodamine B staining
The sulforhodamine B (SRB) assay is a well-investigated assay originally developed by Skehan et al. [33] as a cytotoxicity assay for anticancer-drug screening. SRB binds to surface proteins under acidic conditions and thus can be used to document viability/cytotoxicity.
Twenty-seven hours after the start of the combined transfection and scratch assay, cells were washed once with 1 mL PBS and afterward covered with ethanol (99%). Thereafter, all well plates were stored at −20°C until further investigations. Ethanol was removed, the cells were washed once with tap water, and the plates were then air dried. Cells were covered with 200 μL SRB solution (4% SRB with acetic acid) and incubated while being protected from light. After 30 min of incubation, the SRB solution was removed. The remaining unbound SRB was washed four times with acetic acid solution until fully removed. The bound SRB was resolved with 400 μL 10 mM unbuffered TRIS solution. Finally, absorbance was measured at λ = 565 nm (SRB) and λ = 690 (impurities) and OD690nm was subtracted from OD565nm. The medium-only group was designated as control group (=100% viability).
Statistics
All data are given as mean with standard deviation (SD). Data sets were tested for normality with Kolmogorov–Smirnov test. Normally distributed data were analyzed using repeated-measures analysis of variance with Bonferroni's multiple comparison test to analyze differences between groups. Not normally distributed data were analyzed using a nonparametric test (Friedman test with Dunn's multiple comparison test). All analyses were performed using the statistical software package GraphPad Prism (version 6; GraphPad Software, La Jolla, CA). Statistical significance was defined as P ≤ 0.05.
Results
KGF-mRNA synthesis
A plasmid containing the KGF sequence was used for IVT. After confirmation of successful DNA amplification using gel electrophoresis (Fig. 1A), the DNA was IVT to the corresponding mRNA. KGF-mRNA size and purity was also confirmed on a 1% agarose gel (Fig. 1B).
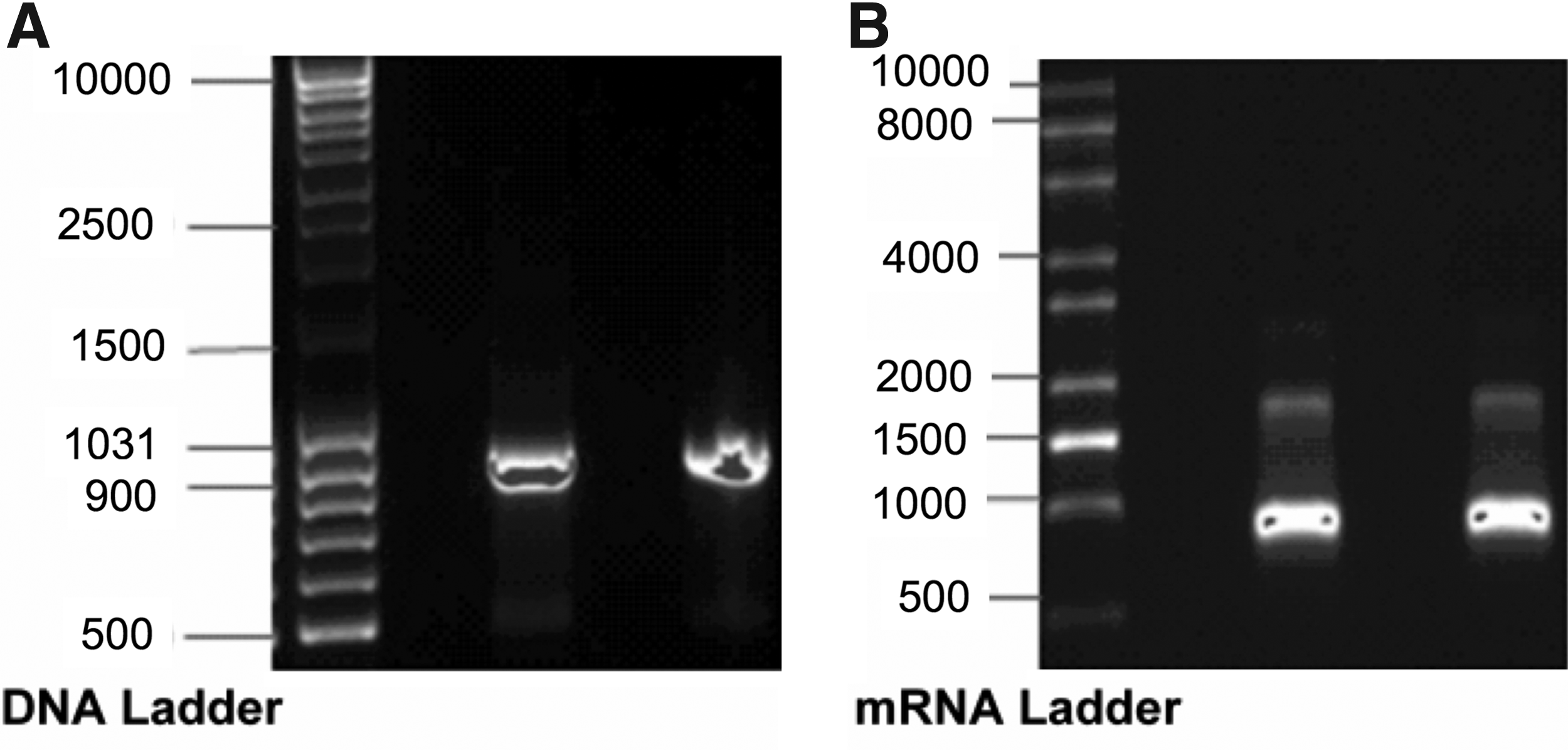
Size and purity of the amplified KGF DNA product
Transfection of cells with KGF-mRNA in vitro
After transfection of HaCat cells, no KGF was detectable after 24 h in the untreated (−31.98 ± 4.38 pg/mL) or Lipofectamine 2000-treated (−3.84 ± 2.03 pg/mL) groups. However, a significantly higher KGF concentration was detected in the group transfected with 0.5 μg (10,992.00 ± 3,329.00 pg/mL; P ≤ 0.0001) or 1 μg (9,007.00 ± 1,585.00 pg/mL; P ≤ 0.0001) KGF-mRNA (Fig. 2A).

The KGF concentration was measured using a specific ELISA after the transfection of HaCat (upper panel) and HFF (lower panel) cells in the supernatant on the first
Similar results were obtained in the HaCat supernatant 2 groups (Fig. 2B). Although control groups again contained no KGF (untreated group: −30.55 ± 2.21 pg/mL, Lipofectamine 2000-treated group: −36.36 ± 2.93 pg/mL), the mRNA-treated groups displayed KGF concentrations of 187.10 ± 207.80 pg/mL or 1,196.00 ± 828.80 pg/mL (P ≤ 0.01).
Transfection of HFF cells also resulted in a significant increase of KGF expression measured in supernatant 1 of cells treated with 0.5 μg KGF-mRNA (6,800.00 ± 3,078.00 pg/mL; P ≤ 0.0001) or 1 μg KGF-mRNA (5,183.00 ± 2,244.00 pg/mL; P ≤ 0.001). Interestingly, the amount of expressed KGF protein in supernatant 2 still increased, that is, 728.30 ± 341.90 pg/mL (P ≤ 0.001) or 747.20 ± 351.90 pg/mL (P ≤ 0.001) in both mRNA treatment groups (Fig. 2C, D).
Similar to the HaCat control groups, no KGF expression was detected in the untreated group (15.33 ± 5.03 pg/mL) or Lipofectamine 2000-treated group (14.67 ± 4.54 pg/mL) of supernatant 1 and in the untreated group (27.29 ± 3.83 pg/mL) or Lipofectamine 2000-treated group (33.36 ± 17.57 pg/mL) of supernatant 2.
Analysis of viability
In addition to KGF expression, cell viability and cell number were determined 48 h after transfection to determine any potential adverse effects of the modified KGF-mRNA (Fig. 3). No significant differences in cell viability were observed in both HaCat (Fig. 3A) and HFF (Fig. 3B)-transfected cells compared with the control groups.
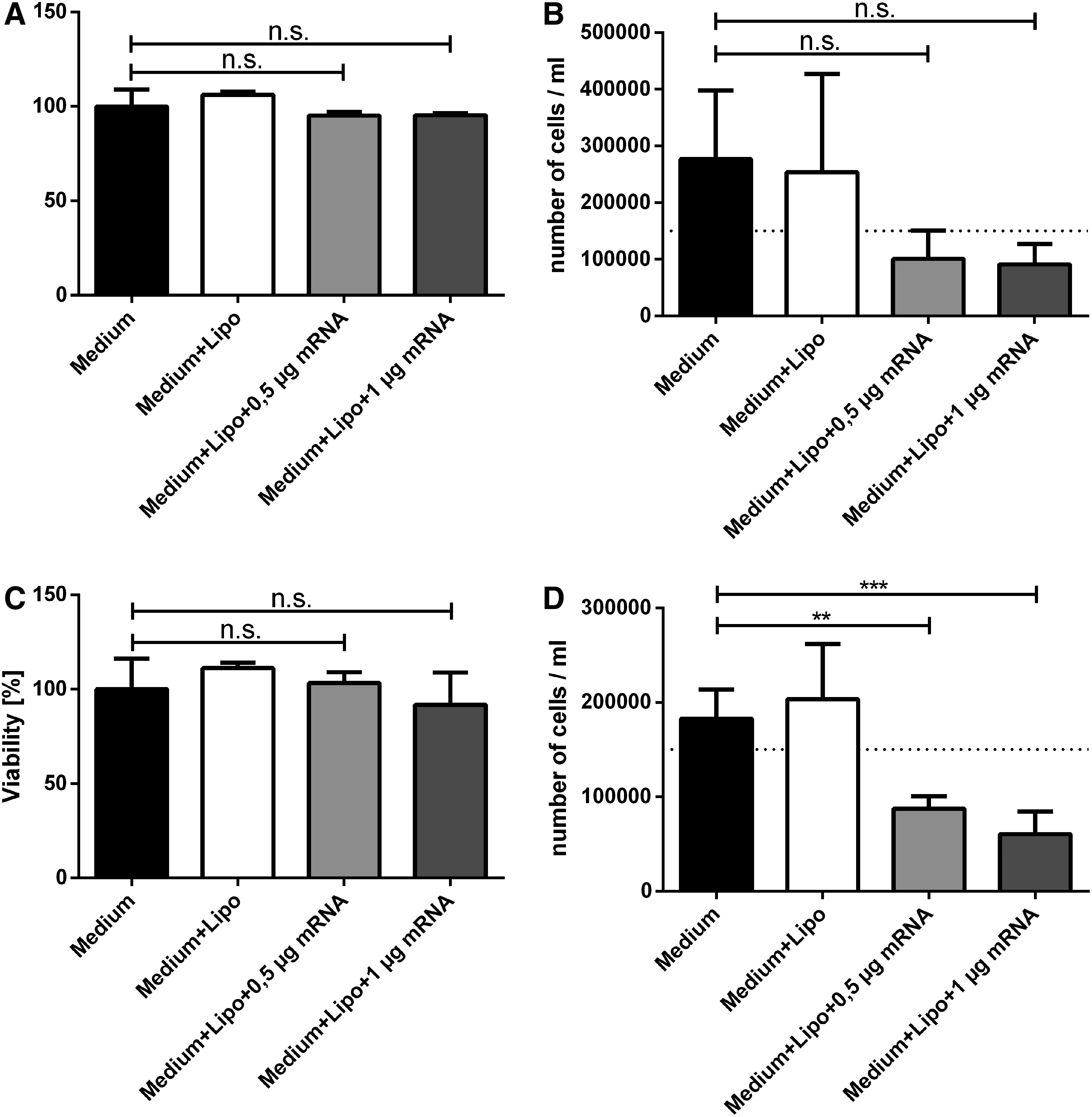
Viability (left column) and cell number (right column) after transfection with HaCat (upper row, n = 4) and HFF (lower row, n = 5) cells. Whereas there was no clear difference detectable between controls and treatment groups concerning cell viability of HaCat
However, differences were visible between the control groups and the mRNA-treated groups with regard to the absolute number of cells measured 48 h after transfection. In HaCat and in HFF transfections, the cell number was lower on average compared with the original seeded cell number. Control groups of both cell lines exhibited a decreased cell number after transfection (HaCat untreated group: 276,250 ± 121,884/mL and Lipofectamine 2000-treated group: 254,000 ± 173,482/mL and HFF untreated group: 182,920 ± 30,828/mL and Lipofectamine 2000-treated group: 203,460 ± 58,537/mL), whereas all treatment groups exhibited a reduction of the cell number after transfection (HaCat 0.5 μg KGF-mRNA group: 100,795 ± 49,784/mL and 1 μg KGF-mRNA group: 91,135 ± 35,706/mL and HFF 0.5 μg KGF-mRNA group: 87,742 ± 12,947/mL and 1 μg KGF-mRNA group: 60,504 ± 24,003/mL).
Analysis of cell migration
To demonstrate that basic conditions in all scratch assays were the same for control and treatment groups, we analyzed the percentage of wound size of the entire image in duplicates at t0 in all images (n = 4 HaCat supernatants 1 and 2; n = 5 HFF supernatants 1 and 2, data not given).
Average sizes of the wounds were similar in the control group (39.68% ± 2.94%) and the treatment groups (0.5 μg KGF-mRNA group: 40.58% ± 2.24% and 1 μg KGF-mRNA group: 40.60% ± 2.19%). The relatively small SD indicates high precision in the technical implementation of scratch assays.
Scratch assays with HaCat supernatants showed a clearly recognizable faster reepithelialization in treatment groups in both scratch assays with HaCat supernatant 1 and HaCat supernatant 2, particularly on day 2 as exemplarily shown in Fig. 4A.
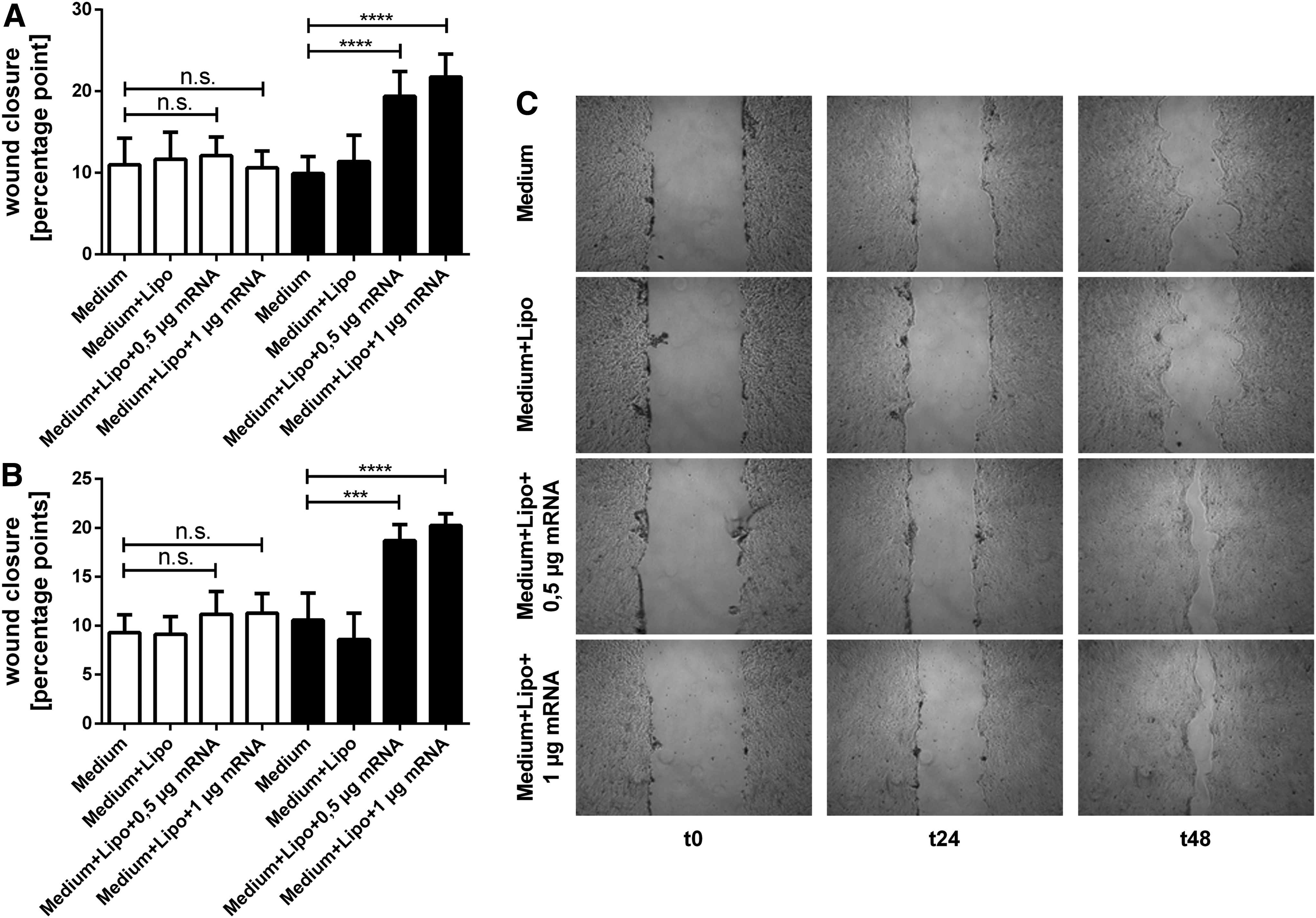
Scratch assay with HaCat supernatants. Reepithelialization after treatment with HaCat supernatant 1
Statistical analysis indicates that there was no significant difference in scratch closure in the scratch assay with HaCat supernatant 1 (Fig. 4B) between control (untreated group: 10.98 ± 3.25 pp and Lipofectamine 2000-treated group 11.67 ± 3.30 pp) and treatment groups (0.5 μg KGF-mRNA group: 12.12 ± 2.28 and 1 μg KGF-mRNA group: 10.61 ± 2.05 pp) on day 1. Treatment groups on day 2 instead showed clear faster reepithelialization (0.5 μg KGF-mRNA group: 19.38 ± 3.06 pp and 1 μg KGF-mRNA group: 21.77 ± 2.80 pp) than controls (untreated group: 9.91 ± 2.08 pp and Lipofectamine 2000-treated group: 11.39 ± 3.21 pp) when treated with HaCat supernatant 1.
Similar results were obtained when cells were treated with HaCat supernatant 2 (Fig. 4C). Again, there were no notable differences in scratch closure on day 1, whereas on day 2 treatment groups exhibited faster reepithelialization (0.5 μg KGF-mRNA group: 18.69 ± 1.66 pp and 1 μg KGF-mRNA group: 20.23 ± 1.22 pp compared with untreated group: 10.57 ± 2.78 pp and Lipofectamine 2000-treated group: 8.59 ± 2.68 pp).
In sum, reepithelialization in the treatment groups on day 2 was approximately as twice as fast as in control groups in both scratch assays with HaCat supernatant 1 and supernatant 2.
As exemplarily shown in Fig. 5A, the scratch assays with HFF supernatants exhibited only slightly faster scratch closure in treatment groups. As previously observed, these differences were the most pronounced on day 2.
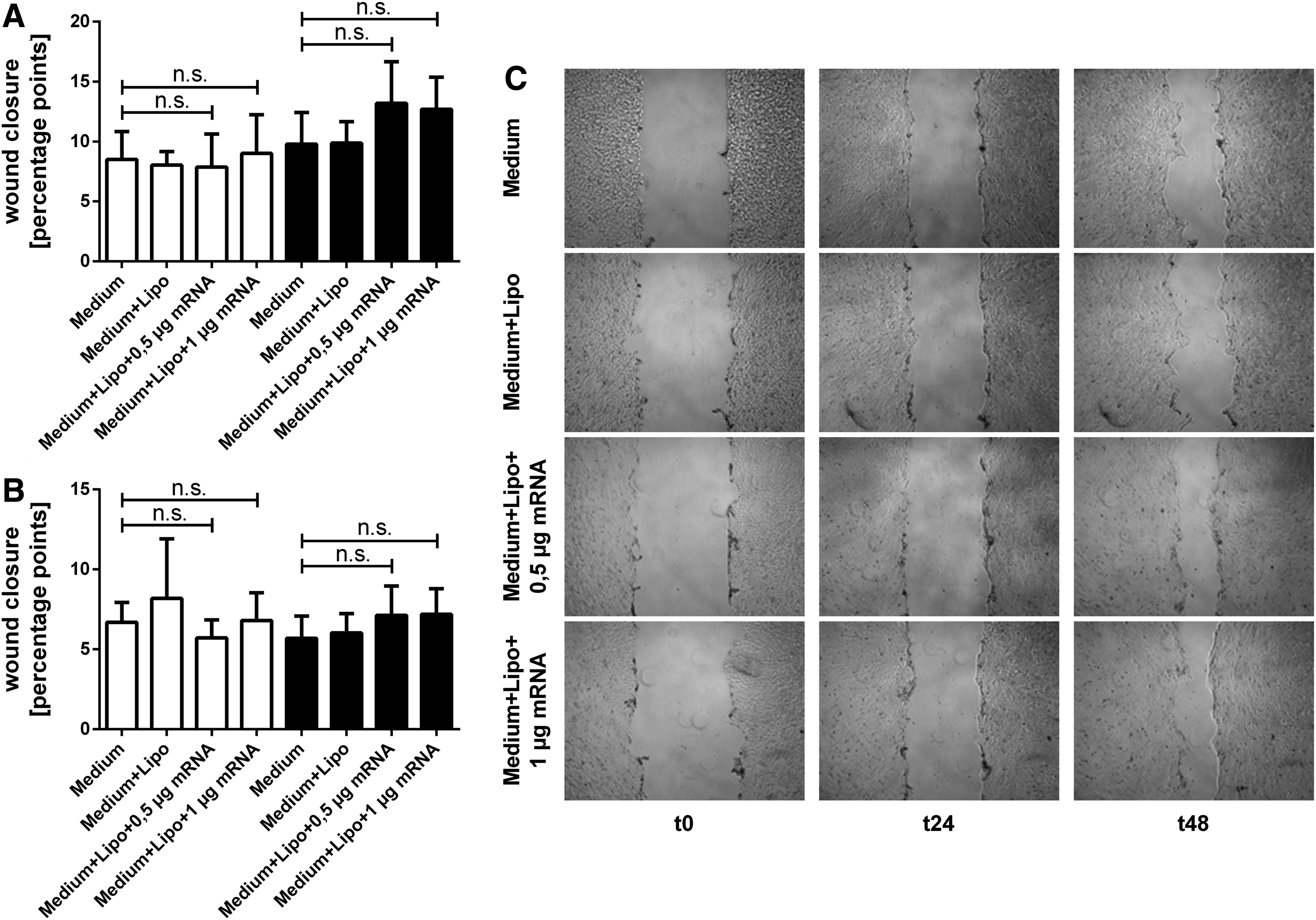
Scratch assay with HFF supernatants. reepithelialization after treatment with HFF supernatant 1
For treatment with HFF supernatant 1, statistical analysis indicates that in the control (untreated group: 8.503 ± 2.328 pp and Lipofectamine 2000-treated group: 8.04 ± 1.14 pp) and in the treatment groups (0.5 μg KGF-mRNA group: 7.88 ± 2.75 pp and 1 μg KGF-mRNA group 9.011 ± 3.24 pp) scratch closure on day 1 was similar. However, on day 2 the treatment groups both showed slightly faster reepithelialization (0.5 μg KGF-mRNA group: 13.20 ± 3.46 pp and 1 μg KGF-mRNA group: 12.68 ± 2.70 pp) than the controls (untreated group: 9.80 ± 2.63 pp and Lipofectamine 2000-treated group: 9.87 ± 1.80 pp) when treated with HFF supernatant 1 (Fig. 5B).
Similar results were observed in scratch assays with HFF supernatant 2 (Fig. 5C): No obvious differences in wound closure became apparent on day 1, but on day 2 treatment groups exhibited a slightly faster reepithelialization than controls (0.5 μg KGF-mRNA group: 5.70 ± 1.39 pp and 1 μg KGF-mRNA group: 6.03 ± 1.21 pp compared with untreated group: 7.13 ± 1.84 pp and Lipofectamine 2000-treated group: 7.20 ± 1.60 pp).
In general, scratch assays with HFF supernatant exhibited slower scratch closure in all groups compared with scratch assays with HaCat supernatants.
After 48 h (scratch assays with HaCat supernatants) or 72 h (scratch assays with HFF supernatants), we evaluated the viability of the HaCat cells (Fig. 6). Neither treatment with HaCat supernatant 1 (Fig. 6A) or 2 (Fig. 6B), nor treatment with HFF supernatant 1 (Fig. 6C) or 2 (Fig. 6D) resulted in significantly different cell viability. Furthermore, no tendency was visible.
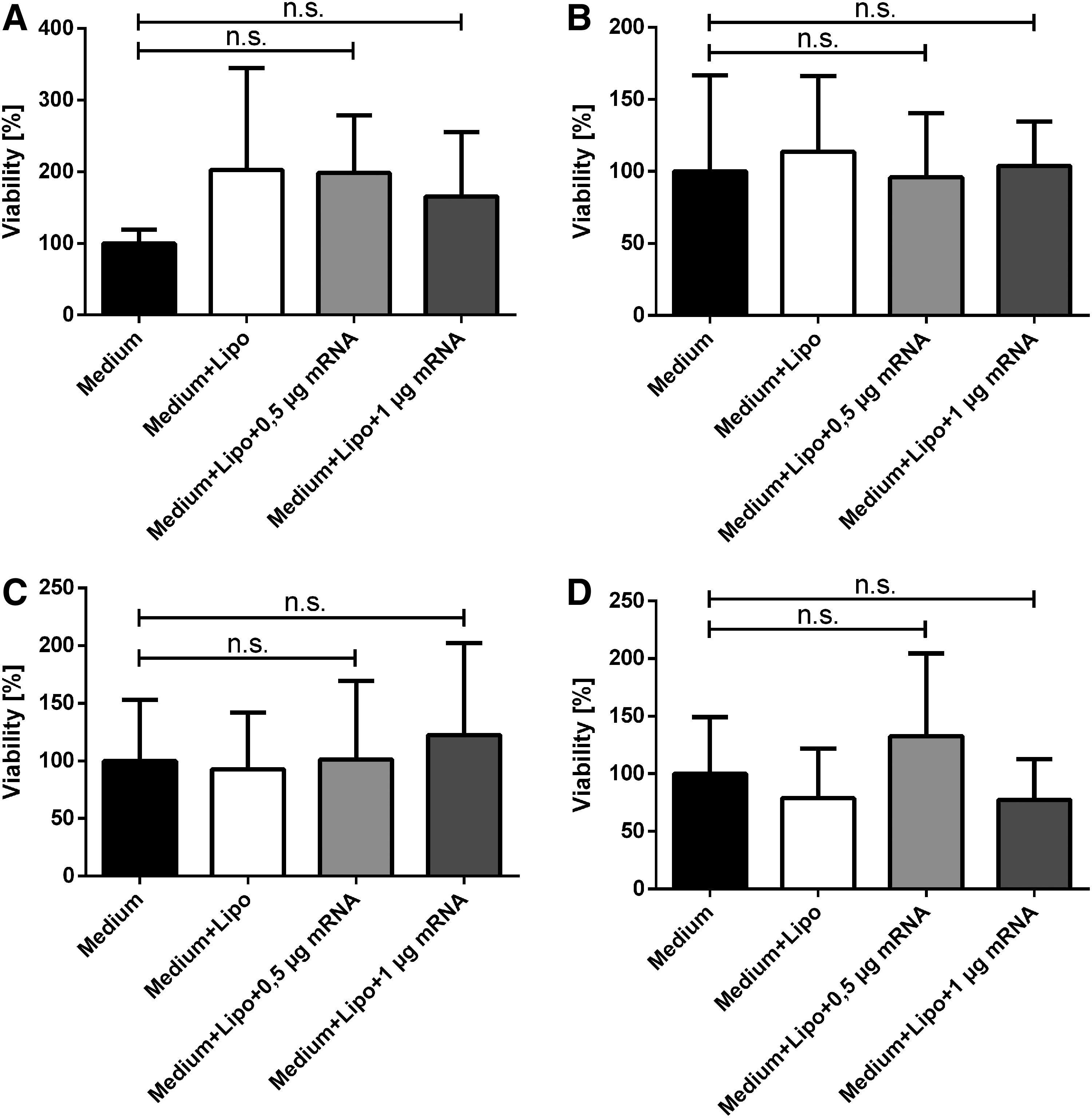
Viability after treatment with supernatants. HaCat supernatants 1
Correlation between KGF concentration and wound healing
In vitro reepithelialization on day 2 (where the greatest differences were detected) and the KGF concentration in the corresponding supernatants showed a positive correlation coefficient for all four scratch assays (Fig. 7). Although this correlation was strong in the scratch assays with HaCat supernatants (scratch assay with HaCat supernatant 1: r = 0.7400 and HaCat supernatant 2: r = 0.5758) (Fig. 7A, B), it was weaker, but nonetheless positive in scratch assays with HFF supernatants (scratch assay with HFF supernatant 1: r = 0.2535 and HFF supernatant 2: r = 0.2113) (Fig. 7C, D).
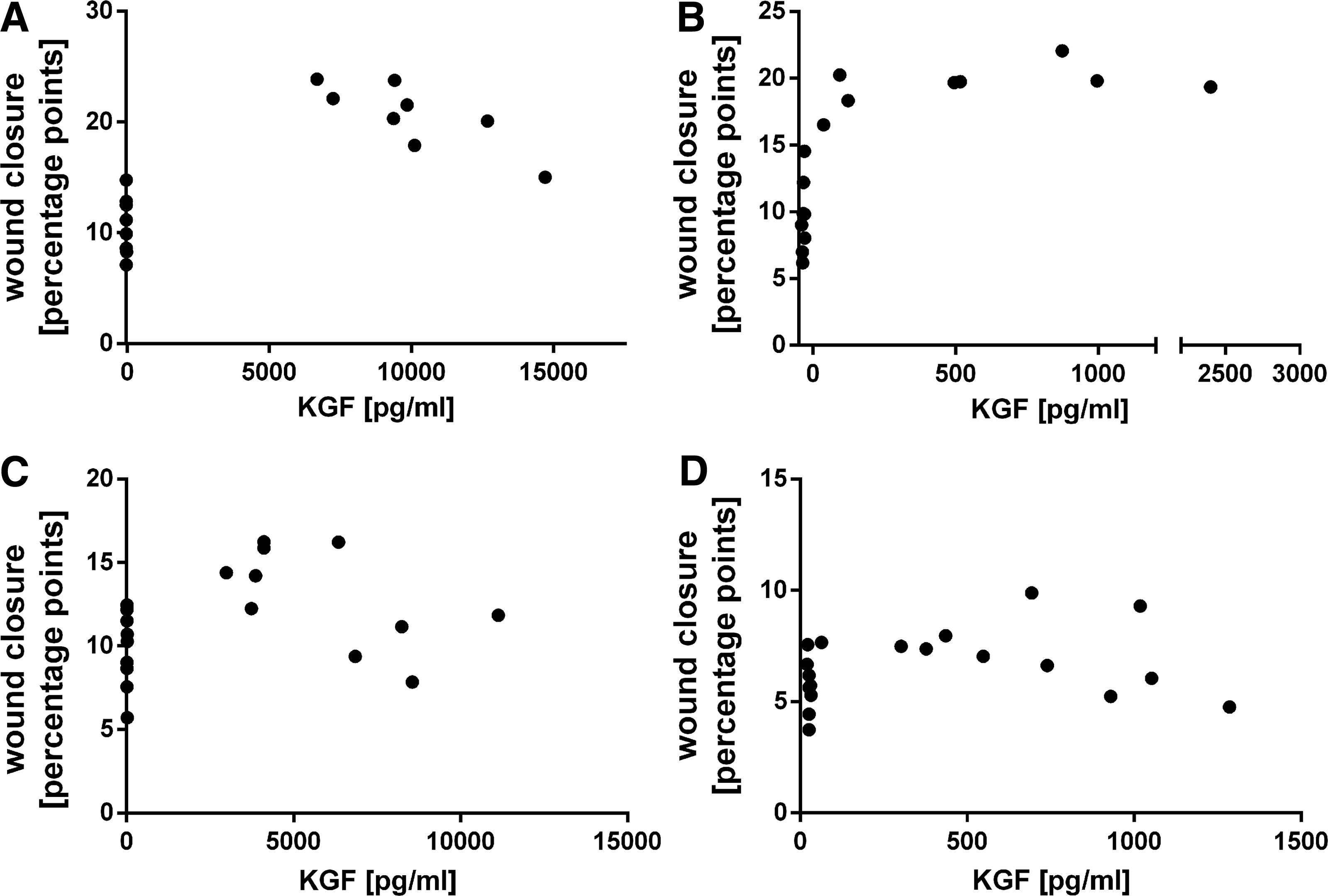
Correlation between KGF concentration and in vitro reepithelialization on day 2 in the scratch assays with HaCat supernatants 1
Combined transfection and scratch assay
Although there was no significant difference in wound closure on day 1, on day 2 KGF-transfected groups exhibited faster wound closure (0.5 μg KGF-mRNA group: 5.87 ± 1.13 pp and 1 μg KGF-mRNA group: 6.22 ± 0.87 pp compared with untreated group: 0.70 ± 1.76 pp and Lipofectamine 2000-treated group: 1.41 ± 0.94 pp). EGFP-transfected groups (0.5 μg eGFP-mRNA group: 2.72 ± 2.37 pp and 1 μg eGFP-mRNA group: 1.580 ± 2.13 pp) showed similar wound closure to control groups (Fig. 8A).
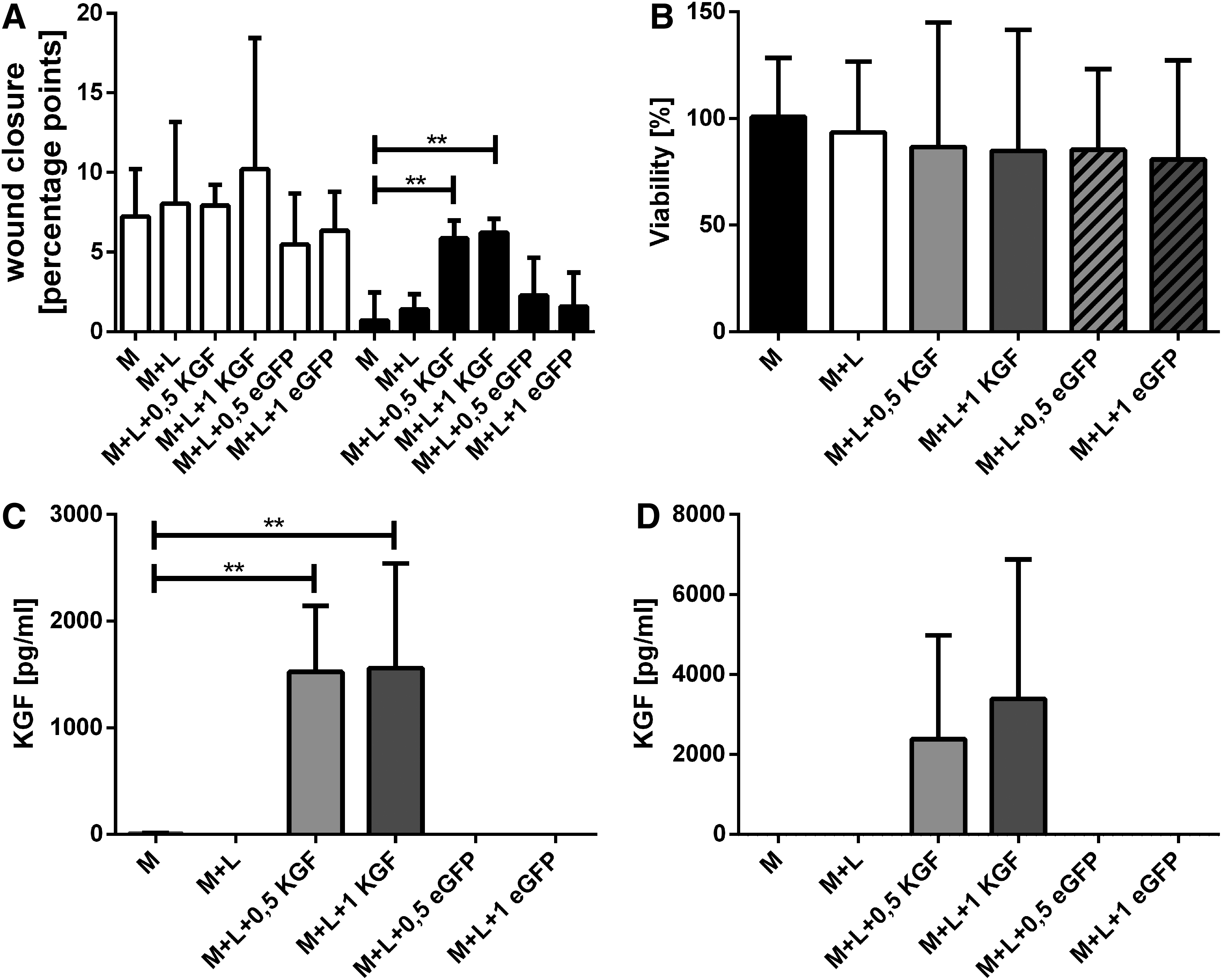
Combined transfection and scratch assay. Analysis of reepithelialization after KGF-mRNA transfection of HaCat cells on day 1 (white column) and day 2 (black column)
No significant difference in cell viability within the different groups was detectable with SRB staining (Fig. 8B).
The KGF concentration in the supernatants of day 1 and day 2 after transfection was analyzed using a specific KGF ELISA (Fig. 8C). In supernatants of day 1 only very low amounts of KGF protein were detectable in the control groups and eGFP groups (0.5 μg eGFP-mRNA group: 1.60 ± 0.26 pg/mL; 1 μg eGFP-mRNA group: 1.56 ± 0.23 pg/mL). When compared with the control group (untreated group: 7.19 ± 5.90 pg/mL), KGF-transfected cells showed significantly higher KGF concentrations (0.5 μg KGF-mRNA group: 1,523 ± 622.1 pg/mL; 1 μg KGF-mRNA group: 1,559 ± 983.5 pg/mL). In addition, on day 2 high amounts of KGF were measured in the KGF-mRNA-treated groups (0.5 μg KGF-mRNA group: 2,384 ± 2,595 pg/mL; 1 μg KGF-mRNA group: 3,390 ± 3,483 pg/mL) (Fig. 8D). There were again no noteworthy amounts of KGF in the supernatants of eGFP-transfected HaCat cells (0.5 μg eGFP-mRNA group: 2.10 ± 0.64 pg/mL; 1 μg eGFP-mRNA group: 1.68 ± 0.24 pg/mL).
Discussion
KGF, which belongs to the fibroblast growth factor family (FGF-7) [34], was first discovered by Rubin et al. [35]. Aside from wound healing, KGF seems to be important in the differentiation of hair follicles [36] and regeneration of cartilage tissue [37]. Previous studies [38–41] demonstrated that KGF could improve healing and thus represents a promising target for further wound healing investigations.
In the 1990s, several growth factor-based products were investigated [42]. In 1999, the recombinant platelet-derived growth factor (rhPDGF) becaplermin was introduced to the market and resulted in improved wound healing in three multicenter, randomized, placebo-controlled, parallel group studies, whereas another study could not show any differences between the becaplermin group and the good ulcer care-alone group [43]. Since 2004, palifermin, a recombinant KGF protein, was available for the prevention of oral mucositis [44], but other indications are already discussed [45].
A series of studies [22,25,46] have focused on the effects of KGF on wound healing in a scratch/migration assay with different experimental settings. Although some studies did not detect a higher keratinocyte migration or scratch closure at the concentrations tested, other studies suggested that KGF (and numerous other cytokines) stimulate keratinocyte migration [47]. Koivisto et al. [25] indicated that a KGF concentration of only 2 ng/mL enhanced keratinocyte migration. In this study, most of the experimental parameters (24-well plate, FCS reduction before scratch) were comparable with those used in our scratch assays.
In this study, we demonstrate that it is possible to induce the expression of KGF in vitro using modified KGF-mRNA. Even keratinocytes, which are not yet known to physiologically express KGF, showed elevated KGF concentrations in the supernatant after transfection. Transfection with our specific mRNA resulted in even higher KGF concentrations that have promoted reepithelialization in previous studies. Furthermore, the used mRNA concentration seems to have an influence on the duration of translation because groups treated with higher KGF-mRNA concentrations exhibited higher expression levels on day 2.
Although we could not locate any previous studies regarding the potential benefits of mRNA therapy on wound healing, some gene therapeutic approaches currently exist with KGF as a target. As the starting point of gene therapy in the biological process of protein synthesis is one step before mRNA therapy, these studies could enable predictions regarding the potential future application of mRNA therapy in wound healing.
The in vivo application of keratinocytes overexpressing KGF resulted in improved wound healing in swine [11]. Marti et al. [12] indicated that KGF expression was increased after electroporative transfection with KGF DNA, and that elevated levels of KGF-mRNA existed in cultured mouse cells. In addition, treated mice showed improved wound healing. Another study demonstrated that topical delivery of KGF DNA strengthens the skin and thickens the epidermis [48]. Improved wound healing was also observed when a KGF plasmid was released from a special scaffold [49]. Wang et al. [50] used adenoviruses for DNA transfection and suggested that this could improve secondary flap necrotic wound healing in an extended animal model. However, as gene therapy with growth factor DNA commonly leads to a permanent increase in growth factor expression resulting in a dedifferentiation and deregulation of the physiological cell cycle, this poses a risk of developing malignancies. As transfection with mRNA is only temporary and does not interfere with the DNA of the treated cells, mRNA therapy therefore seems to be a safer option. Other alternative DNA-based gene therapy strategies also leading to an only transient protein expression are quite ambivalent in case of wound healing. MRNA function initiates directly in the cytosol. Although plasmid DNA (pDNA) also acts in cytosol, it can only be incorporated into cells that are going through mitosis at the time of transfection. This reduces the number of transfected cells in the tissue [51,52]. Specifically, the transfection of tissues with weak mitosis activity, such as cardiac cells, is difficult [53]. In contrast to pDNA, transfection and translation of mRNA occur in mitotic and nonmitotic cells in the tissue [51,54].
Many other growth factors exist that could cause similar improvement in cell migration and enhanced proliferation [47]. These growth factors and cytokines might be additional factors for mRNA-based wound healing therapy. The triggered effects of different growth factors could also be used to induce specific healing processes in various tissues. Therefore, transfection with more “vessel-specific” growth factors could improve ingrowth of vascular grafts, whereas more “skin-specific” growth factors might be used for topical delivery out of a wound dressing in complex wound therapy.
As transfection seems to have toxic effects on the transfected cells, it remains unclear whether KGF-mediated cell proliferation predominates over these toxic effects in vivo. Therefore, we aimed to distinguish between the target cells for transfection and the target cells for growth factors by separating these two steps in our experimental setting to exactly evaluate which effects are caused by the mRNA transfection or by the de novo synthesized KGF.
Nevertheless, in the following experiments, where scratched cells were directly treated with the KGF-mRNA, these groups exhibited a significantly faster wound closure than all control groups on day 2, similar to what was previously observed with scratch assays with supernatants. This indicates that although mRNA seems to have some toxic effects toward the transfected cells, the cell proliferation and migration predominate over the toxic effects. Moreover, eGFP-transfected groups showed similar wound closure to controls indicating that the expressed KGF is responsible for this effect. This is supported by the fact that again only KGF-mRNA-transfected cells showed high KGF concentrations in their supernatants.
As most of the growth factors act in a paracrine manner (which means that the synthesizing cells themselves often are not the target for the growth factors), it could be possible that although a part of the directly transfected cells could be harmed, the other cells and tissue surrounding the transfected area is stimulated and wound healing is induced in vitro and in vivo. However, future in vitro and in vivo studies should aim to discover whether this encouraging new approach can be successfully implemented within in vivo wound healing experiments.
Through investigating the possible cause for toxic effects caused by mRNA transfection as seen in our study, it is thought that one way to reduce this negative side effect is to alter the RNA recognition by Toll-like receptors. It is known that the recognition of RNA by Toll-like receptors [55–58] or retinoid-inducible gene [59,60] can induce an inflammatory process. Physiologically, these mechanisms are directed against virus infections. The implementation of modified nucleosides (pseudouridine and others) resulted in lower inflammatory activation of dendritic cells, as they can no longer identify them as exogenous [61], and in addition increased the translation levels of IVT mRNA [62]. The purification of nucleoside-modified mRNA with high-performance liquid chromatography (HPLC) further eliminates immune activation and improves the translation of nucleoside-modified protein-encoding mRNA [63] that could reduce the minor impurities in the mRNA we observed.
Further modification of the mRNA (eg, modification of the cap structure) could also lead to improved translation of the transfected mRNA [64–66]. These strategies pose promising options for the reduction of toxic side effects and could improve translational efficiency in future studies.
There are already some techniques for topical delivery of mRNA (in case of wound healing we recommend topical application instead systemic delivery). Golombek et al. [67] successfully showed intradermal topical mRNA delivery in an ex vivo porcine model using a microneedle technique. Various other techniques are known for gene delivery in skin, such as subcutaneous injection, electroporation, liquid jet injection, and gene gun delivery [68,69]. Especially the stratum corneum seems to be a strong barrier for macromolecules, such as synthetic mRNAs [68].
As there are already successful approaches of gene therapy with KGF in wound healing, it is likely that mRNA therapy could have similar effects. Besides the application as a wound dressing, this technique could also be applied in skin grafting in the future.
Conclusion
This study demonstrates that it is possible to induce the expression of a specific growth factor by cell transfection using modified mRNA and shows that the amount of KGF protein after transfection is sufficient to promote healing processes. With this technique, it could be possible to achieve longer term elevated concentrations of growth factors in the wound area and in consequence enhance wound healing. Nevertheless, further in vitro investigations and animal studies should be conducted before implementing this technique as a wound therapy.
Footnotes
Author Disclosure Statement
No competing financial interests exist.