
Abstract
Antisense oligonucleotides (ASOs) are classified into gapmer and non-gapmer types according to their chemical modification pattern and mechanism of action. Although gapmer ASOs effectively reduce target RNA expression through intracellular RNase H1, high-affinity gapmer ASOs also have hepatotoxic potential. Non-gapmer ASOs, which are mainly used for pre-mRNA splicing regulation or functional inhibition of microRNA through their steric effects, are also able to inhibit target RNA expression using nonsense-mediated decay. However, it was unknown if they induce high knockdown activity without showing hepatotoxicity. In this study, we investigated the modification pattern of non-gapmer ASOs and show that they have comparable knockdown potential if they have an appropriate melting temperature (Tm) range. We also demonstrated that non-gapmer ASOs show high knockdown effects without inducing hepatotoxicity in the mouse liver. These results indicated that non-gapmer ASOs have the potential to become an alternative inhibitor of target expression with a lower risk of hepatotoxicity.
Introduction
A
The other type of ASO is known as a non-gapmer, which includes fully or partially modified ASOs. Non-gapmer ASOs function by sterically blocking target RNAs without inducing their degradation, and are mainly used for the splicing regulation of pre-mRNAs or the functional inhibition of microRNAs. In 2016, two non-gapmer ASOs, eteplirsen (EXONDYS 51) and nusinersen (Spinraza), were approved by the FDA for the treatment of patients with Duchenne muscular dystrophy and spinal muscular atrophy, respectively [14]. Considering their RNase H1-independent mechanism, non-gapmer ASOs are unsuited to inhibiting the expression of target RNAs, but are expected to show a lower risk of hepatotoxicity than gapmer ASOs. Recently, it was reported that non-gapmer ASOs induce the degradation of target mRNAs using the mechanism of splicing regulation and nonsense-mediated decay (NMD) that is widely preserved among eukaryotes [15]. This study showed that the systemic administration of non-gapmer ASOs targeting mouse Stat3 exon17 caused exon 17 skipping and around 30% knockdown in mouse liver. However, it was unclear if non-gapmer ASOs have the potential for higher knockdown activity without hepatotoxicity.
In this study, we investigated the pattern and variety of sugar modifications in the design of non-gapmer ASOs, including a series of bridged nucleic acids such as 2′,4′-BNA/LNA, amido-bridged nucleic acid (AmNA) [16], BNANC [17], and BNACOC [18,19]. We demonstrated that some non-gapmer ASOs have the potential for comparable knockdown activity to that of gapmer ASOs. We also found that an appropriate melting temperature (Tm) range was more important for non-gapmer ASOs to show high knockdown activity than the steric structure of the sugar modification. Most importantly, we discovered that the systemic administration of AmNA-modified non-gapmer ASOs targeting mouse Stat3 caused high knockdown activity (around 75%–80%) in the mouse liver without inducing hepatotoxicity. This non-gapmer ASO had no knockdown effect in the kidney or lung, but exon skipping was induced in these tissues as well as in the liver. These results suggested that non-gapmer ASOs have the potential to become an alternative inhibitor of target expression with a low risk of hepatotoxicity by making efficient use of an RNase H1-independent mechanism of action.
Materials and Methods
Antisense oligonucleotides
All AmNA-modified ASOs were synthesized and purified by Shionogi Co., Ltd. (Osaka, Japan) as previously described [16, 20]. All other modified ASOs were purchased from Gene Design, Inc. (Osaka, Japan). The structures of sugar modifications and ASO sequences used in this study are shown in Supplementary Fig. S1 and listed in Supplementary Table S1.
Cell culture
Mouse hepatoma Hepa1c1c7 cells were obtained from the American Type Culture Collection (Rockville, MD) and maintained at 37°C with 5% CO2 in Minimum Essential Medium (MEM)α (Gibco, Gaithersburg, MD) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics.
ASO transfection and quantitative reverse transcription-PCR
Hepa1c1c7 cells were seeded in 96-well plates (Corning, Corning, NY) containing 10% FBS/MEMα. After 24 h, ASOs were transfected with Lipofectamine 2000 or 3000 (Invitrogen, Gaithersburg, MD) according to the manufacturer's protocol. After a further 24 h, total RNA was extracted with a CellAmp Direct RNA Prep Kit (TaKaRa Bio, Inc., Shiga, Japan) according to the manufacturer's instructions. Quantitative reverse transcription-PCR (qRT-PCR) was performed with a One Step SYBR PrimeScript PLUS RT-PCR Kit (Takara Bio, Inc.) and analyzed with a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). The primers used in this study were specific for mouse Stat3 (forward: 5′-CAGTGACTCAAAGCCACCTCATTC-3′; reverse: 5′-GGTGGTCAAGGCAAGCAGTTC-3′) and mouse Gapdh (forward: 5′-TGTGTCCGTCGTGGATCTGA-3′; reverse: 5′-TTGCTGTTGAAGTCGCAGGAG-3′). The level of target (Stat3) gene expression was normalized to that of Gapdh.
Analysis of exon skipping by RT-PCR
To confirm Stat3 exon 17 skipping, total RNA was extracted from Hepa1c1c7 cells using an RNeasy Mini kit (Qiagen, Hilden, Germany) and reverse-transcribed into cDNA using a Superscript III First Strand Synthesis kit (Life Technologies, Carlsbad, CA). PCR was conducted using the following primers: mSTAT3-E16-F, 5′-ACTCCTTGCCAGTTGTGGTGATCT-3′ and mSTAT3-E18-R, 5′-TTAGCCCATGTGATCTGACACCCT-3′ with Ex Taq (Takara Bio, Inc.) according to the manufacturer's instructions. PCR products were analyzed on a 10% Tris-borate-EDTA gel stained with GelRed and visualized using ImageQuant LAS 3000 (Fuji Film, Tokyo, Japan).
UV melting experiments (Tm measurements)
UV melting experiments were carried out on a SHIMADZU UV-1800 spectrometer equipped with Tm analysis accessory quartz cuvettes of 1 cm optical path length. The UV melting profiles were recorded in 10 mM sodium phosphate buffer (pH 7.2) containing 10 mM NaCl at a scan rate of 0.5°C/min with detection at 260 nm. The final concentration of each oligonucleotide was 2 μM. The two-point average method was used to obtain Tm values, and the final values were determined by averaging three independent measurements, which were accurate to within 1°C.
In vivo experiment
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of Shionogi Co., Ltd., and all experiments were performed in accordance with the Committee's guidelines. C57BL/6J male mice, 7 weeks old, were purchased from CLEA Japan (Tokyo, Japan) and maintained according to guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International. Each mouse received two subcutaneous injections of 20–180 mg/kg ASO at 4-day interval or five injections of 40–100 mg/kg ASO for five consecutive days. Seventy-two hours after the final administration, mice were anesthetized with isoflurane (Abbott Laboratories, North Chicago, IL) and sacrificed.
For qRT-PCR and RT-PCR, ∼40 mg of liver, kidney, or lung sample was stored overnight in RNA later (Qiagen) at 4°C and then at −80°C. For western blot analysis, ∼100 mg liver was immediately frozen in liquid nitrogen and stored at −80°C. Mouse plasma was collected from the vena cava, and then plasma alanine transaminase (ALT) and aspartate transaminase (AST) levels were measured using the Transaminase CII-test Wako (Wako Pure Chemical Industries, Osaka, Japan), according to the manufacturer's instruction for a 96-well format. Total RNA was isolated from RNA later-treated frozen liver, kidney, or lung samples using an RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. cDNA was synthesized from 1 μg total RNA using the Superscript III First Strand Synthesis kit (Life Technologies), and qRT-PCR was performed with SYBR Premix Ex Taq II (Takara Bio, Inc.) and analyzed with a 7500 Fast Real-Time PCR System (Applied Biosystems). Stat3 mRNA levels were normalized to Gapdh and are presented relative to a saline control. The primers used were the same as for the in vitro assay.
To confirm Stat3 exon 17 skipping, the same cDNA samples were used for PCR with the primer set used for the in vitro assay. PCR products were analyzed on a 10% TBE gel stained with GelRed and visualized using ImageQuant LAS 3000 (Fuji Film) as before. To analyze Stat3 protein levels, liver tissues were homogenized in radioimmunoprecipitation assay buffer (Sigma-Aldrich, St. Louis, MO) with complete protease inhibitor cocktail (Roche, Indianapolis, IN) using a tissue lyzer with 7-mm stainless steel beads (Qiagen). Total protein concentrations were measured with a BCA protein assay reagent kit (Pierce/Thermo Scientific, Rockford, IL) according to the manufacturer's protocol. Aliquots corresponding to 4.2 μg protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (5%–20% gradient gel) and blotted onto polyvinylidene fluoride membranes. The membranes were blocked for 1 h in blocking one (Nacalai Tesque, Kyoto, Japan) and incubated overnight at 4°C in Tris-buffered saline with Tween 20 containing 5% bovine serum albumin and monoclonal rabbit anti-Stat3 antibody (1:1,000; Cell Signaling Technology, Danvers, MA), or for 1 h at room temperature in blocking one containing monoclonal mouse anti-β-actin (1:1,000; Sigma-Aldrich). Subsequently, the membranes were incubated for 1 h with horseradish peroxidase-conjugated antibodies (1:3,000; (Amersham Biosciences, Buckinghamshire, United Kingdom). Stat3 and β-actin bands were visualized using the ECL Prime kit (Amersham Biosciences) and ImageQuant LAS 3000 (Fuji Film).
Statistical analyses
Statistical comparisons were made using Student's t-tests. *P < 0.01 or **P < 0.001 was considered to be statistically significant.
Results and Discussion
Gapmer and non-gapmer ASOs show comparable knockdown in vitro
Exon 17 skipping of mouse Stat3 pre-mRNA produces a premature termination codon in exon 18, which induces NMD, resulting in Stat3 mRNA degradation (Fig. 1A). Indeed, fully 2′-MOE-modified ASOs designed in the region of 17a or 17b within exon 17 cause exon 17 skipping and Stat3 knockdown [15], while 2′,4′-BNA/LNA-incorporating non-gapmer ASOs, known as splice-switching oligonucleotides (SSOs), induce exon skipping using their high binding affinity for target RNAs [21]. Therefore, we expected to improve the knockdown activity of non-gapmer ASOs targeting Stat3 exon 17 using bridged nucleic acids such as 2′,4′-BNA/LNA or AmNA.
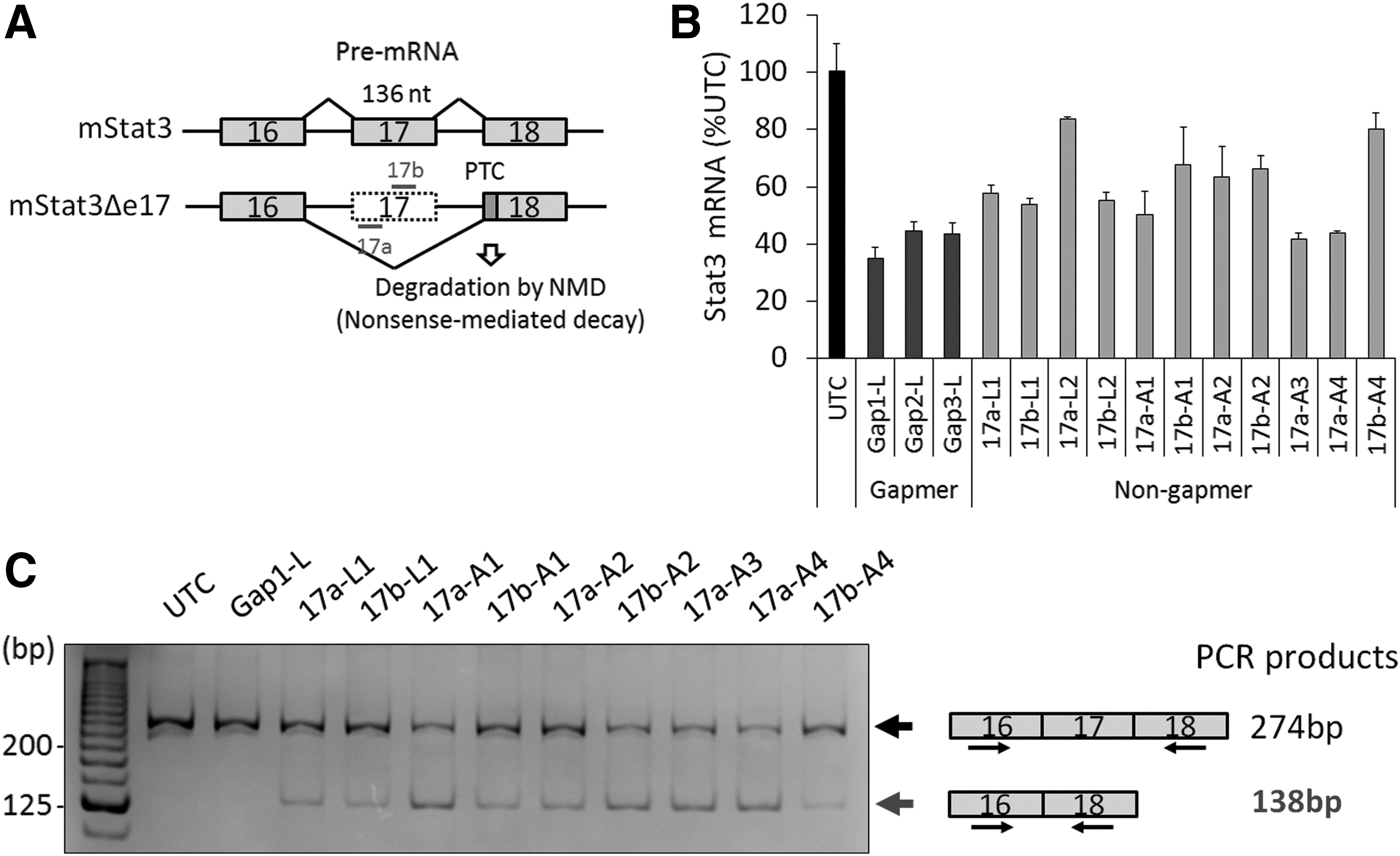
RNA reduction by non-gapmer ASO in vitro.
Based on the ASO sequences previously reported [15]), we designed a series of non-gapmer ASOs targeting Stat3 exon 17 incorporating bridged nucleic acids such as 2′,4′-BNA/LNA or AmNA (Supplementary Table S1). To compare the knockdown activity of non-gapmer ASOs with that of gapmer ASOs, these ASOs were transfected into mouse hepatoma Hepa1c1c7 cells, and the relative expression level of Stat3 mRNA was analyzed by qRT-PCR. Some non-gapmer ASOs such as 17a-A3 or 17a-A4 showed comparable knockdown activity with gapmer ASOs such as Gap1-L, Gap2-L, and Gap3-L (Fig. 1B).
Next, to confirm the mechanism of exon 17 skipping, we analyzed the RNA products by RT-PCR. Exon 17-skipped bands were detected in cells transfected with non-gapmer ASOs, but not in those transfected with gapmer ASOs (Fig. 1C). These results indicate that exon 17 is skipped by non-gapmer ASOs, and then, exon 17-skipped Stat3 mRNAs are degraded by NMD. The knockdown activity also did not seem to depend on the number of modifications. For example, 17a-A4 with 11 AmNA modifications showed similar activity to 17a-A3 with eight AmNA modifications. Because 2′,4′-BNA/LNA-modified SSOs need to have a favorable melting temperature to show efficient exon skipping [21], AmNA-modified non-gapmer ASOs may also require an optical binding affinity to show a high knockdown activity through NMD.
Non-gapmer ASOs require optical Tm values to achieve high knockdown activity
To investigate the relationship between knockdown activity and the complementary RNA binding affinity of AmNA-modified non-gapmer ASOs, we analyzed the Tm values of non-gapmer ASOs with complementary RNAs by UV melting experiments (Fig. 2A). Non-gapmer ASOs having a Tm of 60°C–75°C showed comparable knockdown activity to gapmer ASOs, but non-gapmer ASOs having a Tm of more than 80°C had a relatively low knockdown activity. We also found that non-gapmer ASOs (17a-A3) had comparable dose dependency to gapmer ASOs (Gap1-A) in Hepa1c1c7 cells (Fig. 2B). These results suggested that non-gapmer ASOs potentially have comparable knockdown activity to gapmer ASOs, which does not correlate with their binding affinity to target RNAs.
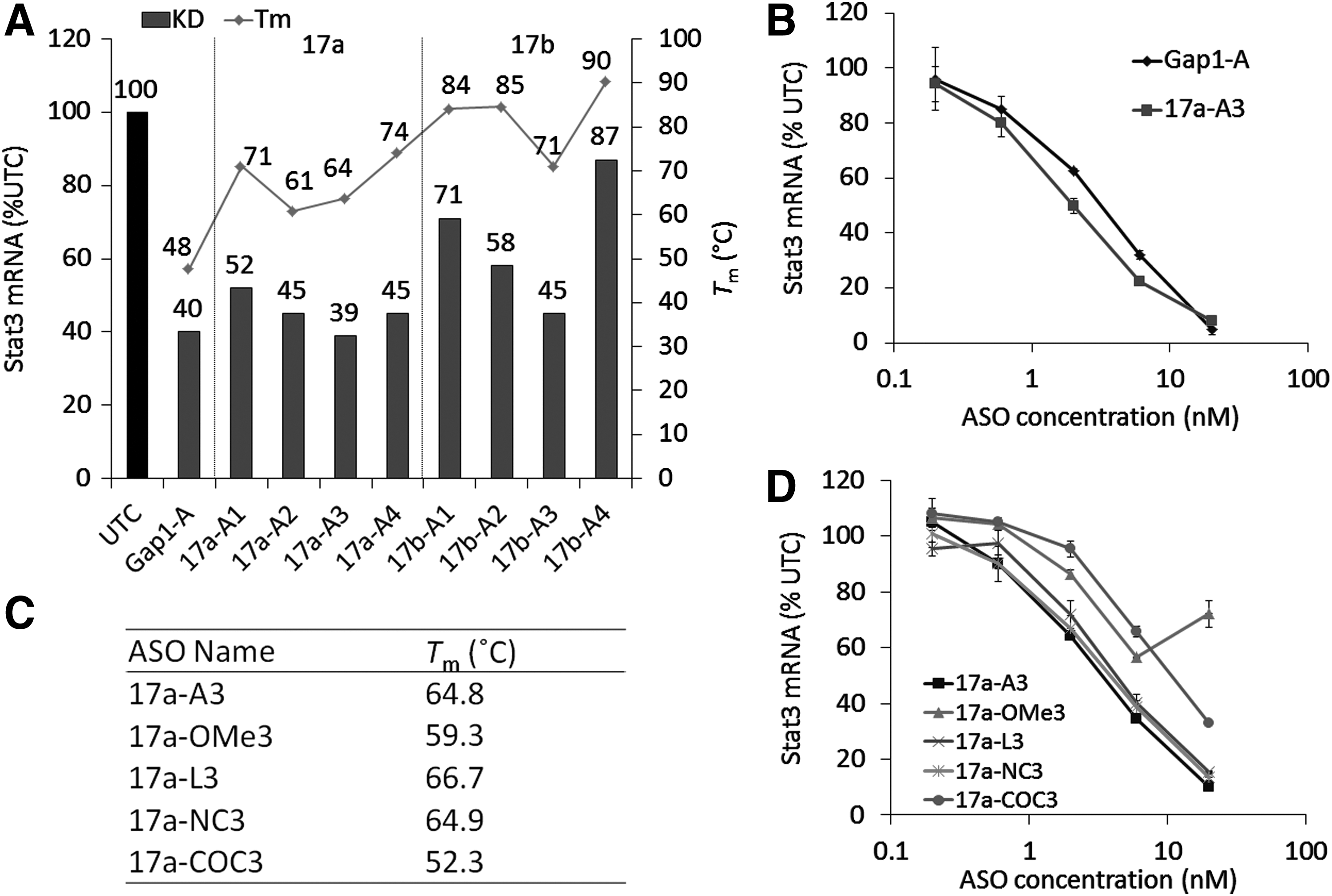
Knockdown activity and Tm value of non-gapmer ASOs.
Next, because non-gapmer ASOs work through steric blocking, we hypothesized that the steric structure, in the form of differences in sugar conformation, may affect the activity of non-gapmer ASOs as well as the Tm value. We therefore prepared ASOs with different Tm values or sugar conformations (Fig. 2C and Supplementary Fig. S1), and compared their knockdown activities (Fig. 2D). Compared with AmNA-modified ASOs (17a-A3), 2′-OMe (17a-OMe3)- or BNACOC (17a-COC3)-modified ASOs, which have lower Tm values than other ASOs, showed lower activity. However, 2′,4′-BNA/LNA (17a-L3)- or BNANC (17a-NC3)-modified ASOs, which have similar Tm values to 17a-A3, showed comparable knockdown activity. These results suggested that non-gapmer ASOs require an appropriate Tm range to show high knockdown activity and the difference in Tm value is more critical for the knockdown activity of non-gapmer ASOs than the steric structure of the sugar modification.
Non-gapmer ASOs induce a high knockdown effect without showing hepatotoxicity in the liver
Although high binding gapmer ASOs show high knockdown activity in vivo, they also have hepatotoxic potential mainly because of their off-target effect induced by RNase H1 [12,13]. Because non-gapmer ASOs showed comparable knockdown activity in vitro without using RNase H1, we next investigated whether they induced high knockdown activity without showing hepatotoxicity in the mouse liver. Non-gapmer ASOs (17a-A3 and 17a-A4) or a gapmer (Gap1-A) ASO were subcutaneously injected into mice at 20 or 40 mg/kg twice, and the expression levels of Stat3 mRNA in the liver and ALT and AST levels in the plasma were analyzed 7 days after the first administration (Fig. 3A). Gap1-A showed around 70% knockdown at 20 mg/kg and more than 90% knockdown at 40 mg/kg, while 17a-A3 showed no significant knockdown at 20 mg/kg, but around 50% knockdown at 40 mg/kg; 17a-A4 showed no significant knockdown at either dosage. Considering that both non-gapmer ASOs showed comparable knockdown to Gap1-A using the transfection reagent in vitro, the difference in in vivo activity may be caused by differences in pharmacokinetics and/or intracellular kinetics among ASOs. Aside from knockdown activity, plasma ALT and AST levels as hepatotoxic makers were not increased in the mice injected with 17a-A3 or 17a-A4.
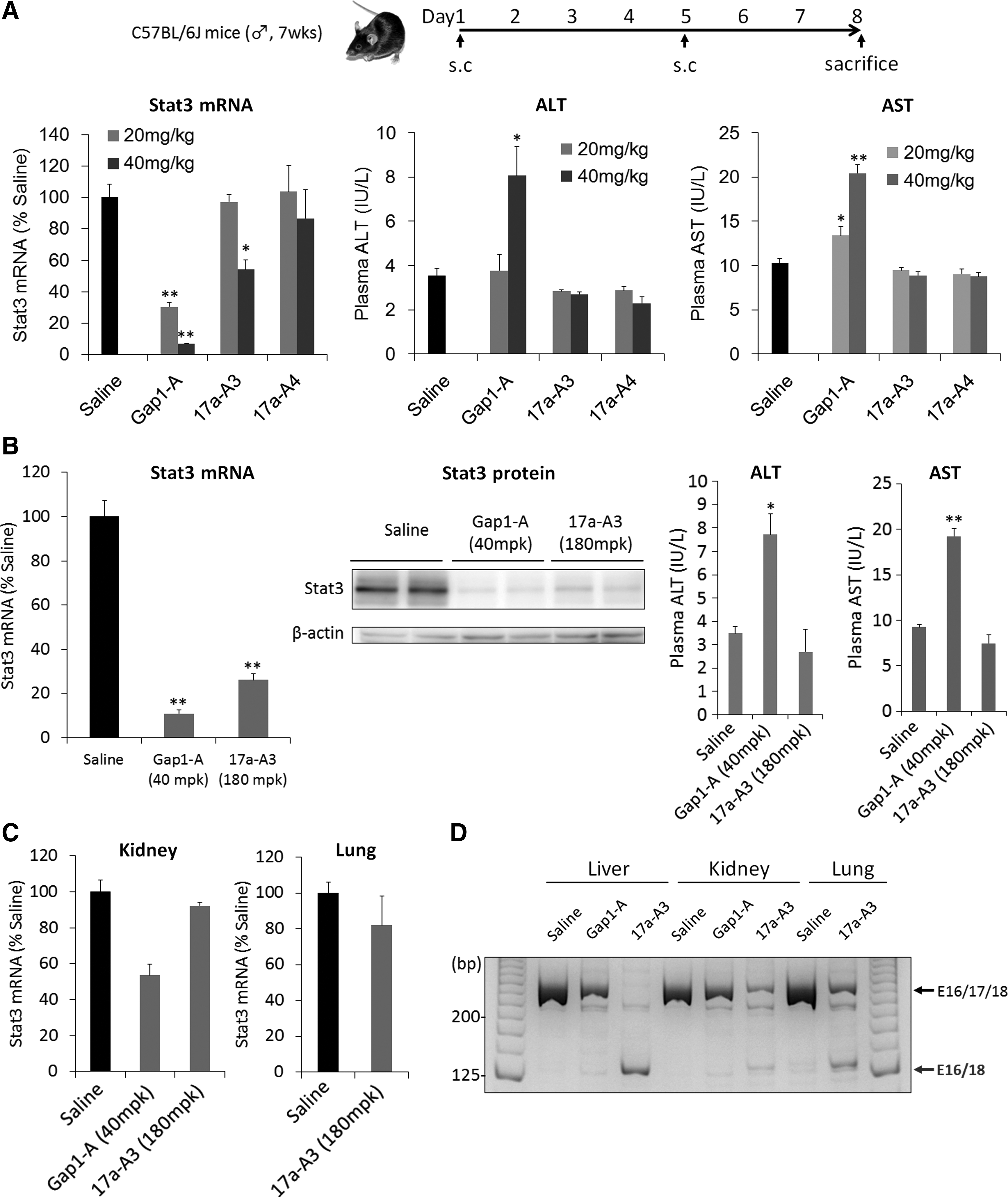
RNA reduction and hepatotoxic potential of non-gapmer ASOs in mice.
Next, to investigate whether non-gapmer ASOs showed higher knockdown activity in vivo, we increased the dosage of 17a-A3 to 180 mg/kg (Fig. 3B and Supplementary Fig. S2) or the frequency of administration (40 or 100 mg/kg for five consecutive days) (Supplementary Fig. S3); 17a-A3 showed around 75% knockdown activity of Stat3 mRNA expression at 180 mg/kg twice or more than 80% knockdown activity at 100 mg/kg five times. We also confirmed the knockdown of Stat3 at the protein level (Fig. 3B). Importantly, ALT and AST levels showed no increase even with two to five injections at the higher dosage (Fig. 3B and Supplementary Fig. S4). These results suggested that non-gapmer ASOs show high knockdown activity without inducing hepatotoxicity in the liver. We also investigated the expression levels of Stat3 in other tissues such as the kidney and lung of the same mice injected with 180 mg/kg of 17a-A3; 17a-A3 did not induce high knockdown in the kidney or lung (Fig. 3C), but exon 17-skipped bands were detected in these tissues as well as in the liver (Fig. 3D). These results indicated that exon skipping was induced in the kidney, lung, and liver, but that more efficient delivery is required to achieve knockdown activity in these tissues.
To demonstrate that non-gapmer ASOs induce knockdown effect without showing hepatotoxicity in the liver, we further designed a different series of non-gapmer ASOs targeting Stat3 exon 6 or 7 by incorporating AmNA into the sequences reported before [22] (Supplementary Table S1). We confirmed that these non-gapmer ASOs also showed various activities in vitro depending on their Tm values (Supplementary Fig. S5). We next investigated whether they induced high knockdown activity without showing hepatotoxicity in the mouse liver. All of these ASOs showed neither hepatotoxicity nor knockdown activity at 40 mg/kg twice (Supplementary Figs. S6 and S7).
In conclusion, while further experiments are required to improve the pharmacological and pharmacokinetic profiles of non-gapmer ASOs, we found that they have a comparable knockdown potential to gapmer ASOs using exon skipping and NMD mechanisms. We also demonstrated that non-gapmer ASOs greatly reduce the expression of target mRNA without showing hepatotoxicity in vivo. In the future, non-gapmer ASOs may be utilized as an alternative inhibitor of gene expression with low hepatotoxic potential by making efficient use of the RNase H1-independent mechanism.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.